孟德尔随机化的良好实践
——孟德尔随机化分析的常见设计、关键挑战及优化
2023-12-19张国燕
王 晶 张国燕 程 杉
(首都医科大学基础医学院医学遗传学与发育生物学学系,北京 100069)
2022年是被称为“现代遗传学之父”的格雷戈尔·孟德尔(Gregor Mendel)诞辰200周年,他通过研究发现的分离规律及自由组合规律,作为遗传学的三大基本规律之二,奠基了整个现代遗传学。1986年,Katan[1]基于孟德尔发现的自由组合规律的思想,提出了一种遗传流行病学研究方法,即孟德尔随机化分析(Mendelian randomization analysis,MR),其原理基于所有的DNA在减数分裂时,遗传变异(genetic variants, G)随机独立地组合,因此,对于某个性状(如:乙醇脱氢酶的酶活性),人群可以依据与其表达水平直接相关的遗传变异(如:ALDH2基因的rs671多态)的基因型进行分类[如:AA(正常酶活性)、AG(50%酶活性)及GG(无酶活性)3组]),这种大自然的随机化设计恰巧类似遗传流行病学研究中常用的随机对照试验(randomized controlled trial, RCT)。
因此,如果某个遗传变异与感兴趣的暴露因素(X)相关,且与结果(Y)无关,那么该遗传变异可以作为“工具变量”(instrumental variable, IV)来代表暴露因素,从而推断暴露因素对结果的因果影响[2-3]。在观察性研究中,因果方向往往不明确,即X是否导致Y或Y是否导致X,而MR方法通过将遗传变异作为IV,更好地帮助研究人员确定因果方向,从而解决观察性研究中常见的混杂和反向因果问题,得出更接近因果关系的结论[4]。
近年来,MR分析在医学研究中的应用加速发展,得益于以下几个方面:首先,IV选择自由度的提升,IV与X相关度越高,因果估计越可靠。大规模基因组数据的公开数据库使得全基因组关联分析研究(genome-wide association studies,GWAS)及通过各种高通量组学技术获得的数量性状位点(quantitative trait locus,QTL)[表达数据(expressin QTL, eQTL),宏基因组数据(microbial environmental genome QTL,mbQTL)及甲基化数据(methylation QTL,mQTL)等]研究对基因组变异认知程度的飞跃,显著扩展了与表现型相关强IV的选择[5];第二,随着全球范围内大规模队列研究和生物银行的建立,越来越多的人群遗传数据及临床数据可供研究人员使用,这有助于增加MR分析的统计功效和可靠性[6];第三,研究人员不断改进MR分析的统计方法,显著提高了因果推断的准确性[7]。
本文将从MR分析的基本假设及常见设计类型,MR分析在具体实施方案中的关键挑战及MR分析的应用前景等几个方面进行阐述。
1 MR分析的基本假设及常见设计类型
MR分析的根基是由遗传变异作为IV,而有效的IV需同时满足以下3个关键假设定义[8-9]:
(1)相关性假设(relevance assumption):即相关性,遗传变异G必须与感兴趣的风险因素X相关联。这意味着这些变异可以影响暴露因素的变异。这个假设的合理性取决于G是否真的与X相关,以及G对X的影响程度。
(2)独立性假设(independence assumption):即独立性,遗传变异G不能与Y存在共同的原因。换句话说,G对Y的影响不能通过其他因素介导。这是为了避免混杂的影响,确保G对Y的影响是直接的。
(3)排除限制假设(exclusion restriction assumption):即排他性,遗传变异G对Y的影响必须仅通过X进行介导。即G不能直接影响Y,而必须通过其关联的X来影响Y。这个假设的合理性确保了遗传变异作为工具变量的有效性。
MR分析发展至今,有多种不同的设计类型,同类问题的多次MR研究对比能更好地阐述MR实施方案的发展和革新,如以探究心血管疾病——冠状动脉粥样硬化性心脏病(coronary artery disease,CAD)、心肌梗死(myocardial infarction,MI)等的风险因素而采用多次不同MR分析方法的研究为例:
单阶段MR(one stage Mendelian randomization):为最早的MR,研究通过假设G-X关联,同时G-Y关联,推测X与Y关联,类似“黑盒算法”,研究中使用一两个已知的遗传变异来作为IV,通过MR分析进行因果推断,研究[10]利用一个遗传变异,KIV-2 基因重复,作为IV,该遗传变异重复次数升高,与脂蛋白(a)水平降低相关,同时该遗传变异重复次数升高与MI发生的风险降低也相关,由此得出了遗传数据支持脂蛋白(a)水平升高与MI风险增加之间的因果关系的推论。然而,这种方法由于因果关联的效应大小无法用数值估计而未能广泛应用。
单样本MR(one sample Mendelian randomization):为对于同一数据集的进阶研究方法,假设一个遗传变异G与某个特定的表型特征暴露X相关联,那么该遗传变异G也应该与该表型特征的结果Y相关联,利用最小二乘法回归模型(two-stage least squares,2SLS)统计分析方法进行计算,提供因果推断的依据。如Patrick等[11]研究,通过关联分析确定的LDLR基因内单核苷酸多态性(single nucleotide polymorphism, SNP) rs2228671的T等位基因可降低低密度脂蛋白胆固醇(low density lipoprotein-cholesterol,LDL-C)水平(拥有0,1,2个T等位基因的LDL-C水平分别为:基线,-3 mg/dL,-6 mg/dL),同时,该等位基因与CAD的风险显著降低相关。通过逻辑回归及MR模型计算,表明LDLR基因座的遗传变异与LDL-C的变化以及CAD的风险之间存在功能性联系。但是,单样本MR仅限于单个样本,IV的选择范围比较有限,且因果关系仅来自同一数据集,容易受弱工具偏倚(weak instrument bias)[12]、水平多效性(horizontal pleiotropy)干扰影响。
两样本MR(two sample Mendelian randomization):以往MR分析常用的外部数据集大多来自Meta分析,但随着技术的发展和遗传数据的增加,尤其是随着GWAS成为探索基因与疾病关联的主要方法之一,大量由GWAS研究产生的SNP及拷贝数变异(copy number variation,CNV)等作为潜在IV的强大数据来源,利用这些SNP或CNV与表型特征(例如暴露因素X或疾病结果Y)之间的关联来推断因果关系[13-14]。而且,大量的GWAS研究数据结果的共享及全球协作组的建立,打破了单样本MR需要在同一样本中同时测量X(暴露因素)及Y(疾病结果)才可进行分析的短板,研究人员可以利用分别来自相似人口背景的两个不同数据集,一个用于分析GWAS数据中G和X(暴露因素)之间的关联,另一个用于分析G和Y(疾病结果)之间的关联,并通过样本量优势及优化的统计分析方法保障更好的因果估计及敏感性分析,这种“借力打力”的研究方法被称为MR。如在观察实验[15]提示维生素 E 对心脏有保护作用,而干预试验未能证实其有益作用,甚至一些研究[16]报告了维生素 E 补充剂对CAD的不利影响。Wang等[17]基于GWAS研究的数据进行了一项两样本MR研究,以调查维生素 E 与 CAD 风险之间的因果关系。根据GWAS结果,rs964184、rs2108622 和 rs11057830 作为与暴露“维生素E”关联的IV;而与CAD/MI及LDL-C/三酰甘油(triglyceride,TG)/总胆固醇(total cholesterol,TC)关联的IV则选自大型生物数据库“冠状动脉疾病全基因组验证和Meta分析与冠状动脉疾病遗传学联盟”[即CARDIoGRAMplusC4D: Coronary Artery Disease Genome Wide Replication and Meta-analysis (CARDIoGRAM) Plus the Coronary Artery Disease (C4D) Genetics]及“国际脂遗传学联盟”(即Global Lipids Genetics Consortium),研究结果显示,维生素E的摄入增高会同时增加LDL-C、TG和TC的浓度,且较高的维生素 E 可能会增加 CAD/MI 的风险。两样本MR也是在目前大数据背景下最常见的MR设计类型,但是要注意的是,由于两样本MR需要使用外部数据,可能会引入选择性偏倚,如使用GWAS研究结果作为IV而产生的胜利者效应(Winner’s Curse),导致IV与X之间的关联被高估,而因果关联被低估[18]等问题。
双向MR(bidirectional Mendelian randomization,BMR):在观察性研究中,因果关系可能是双向的,即X可能导致Y,同时Y也可能导致X,排除反向因果也是在单样本MR及两样本MR研究中的一个弱势。与传统的两样本MR相比,BMR分析方法可以解决潜在的“因果交织”问题,该方法的本质是两样本MR的变体,专门用于评估两个潜在相互关联的因素之间的因果关系。研究方案是同时在两个方向分别进行两样本MR分析,试图确定因果关系的方向,从而避免了反向因果关系所引起的混淆,更全面地理解因果关系。如一些观察性临床研究提示CAD和房颤(atrial fibrillation,AF)相互加重,Tao等[19]通过双向MR的方法评估CAD和AF之间的因果关联,根据结果提示CAD患者与AF风险升高相关,而在AF患者中却未发现与CAD风险的因果关联。
两阶段MR(two step Mendelian randomization,TSMR):在因果推断中有一个重要的概念,“Collider偏差”(Collider bias),指的是当一个变量被作为中介因素或共同因素同时影响两个其他变量时,可能引入虚假的相关性或偏倚,如“冰淇淋消费与溺水事件增加相关”,但实际上并不是冰淇淋与溺水之间有真正的因果关系,而是在夏季时,人们更容易消费冰淇淋和游泳,从而引发了Collider偏差。针对这个问题,两样本MR的另一个变体——TSMR可以用于评估探讨中介变量是否介导了暴露X对结果Y的影响,不仅仅局限于单一因素与某一结果的关联,适用于寻找多个因素之间的复杂关系,通过“拆包”的方法推测暴露X到结果Y的发生机制。如Lin等[20]研究采用了两阶段MR方法,以血糖和血脂风险因素为中介变量,确定血糖和血脂风险因素在体质量指数(body mass index,BMI)对CAD的影响中是否有介导作用。研究分别分析了BMI对CAD、2型糖尿病、空腹血糖、胰岛素、糖化血红蛋白(hemoglobin A1c, HbA1c)、LDL-C、高密度脂蛋白胆固醇(high density lipoprotein-cholesterol,HDL-C)和TG等血糖和血脂风险因素的影响,以及血糖和血脂风险因素与CAD的因果关系。研究结果显示,升高的TG浓度和糖代谢不良可能介导了BMI对CAD的影响。
以上5种MR分析就是目前主流的MR分析类型的发展趋势,每种设计类型都有其优势和值得改进的地方。单阶段 MR 简单易行,适用于单个暴露因素和单个结果变量的情况,但可能受到IV假设不满足和遗传变异的限制。单样本 MR 操作简便,无须外部数据,适用于单个暴露因素和单个结果变量,但更容易受到弱工具偏倚、水平多效性干扰等影响。两样本 MR 提供更丰富的数据来源选择,能够评估因果关系在不同群体中的一致性,更好地控制水平多效性问题,但外部数据的引入,可能引入选择性偏倚。双向 MR 提供更全面的因果关系信息,可以支持因果路径的双向验证,但需要同时满足两个方向的IV假设。两阶段 MR 支持多个因果关系的同时评估,可以发现复杂的因果网络,但IV的有效性受到遗传变异的强度和频率影响,同时需要更多的数据和统计分析,结果的解释更具挑战。表1中将上述5种MR主要类型的基本原理、分析方法、优势、弱势及潜在改进方向进行了归纳汇总。
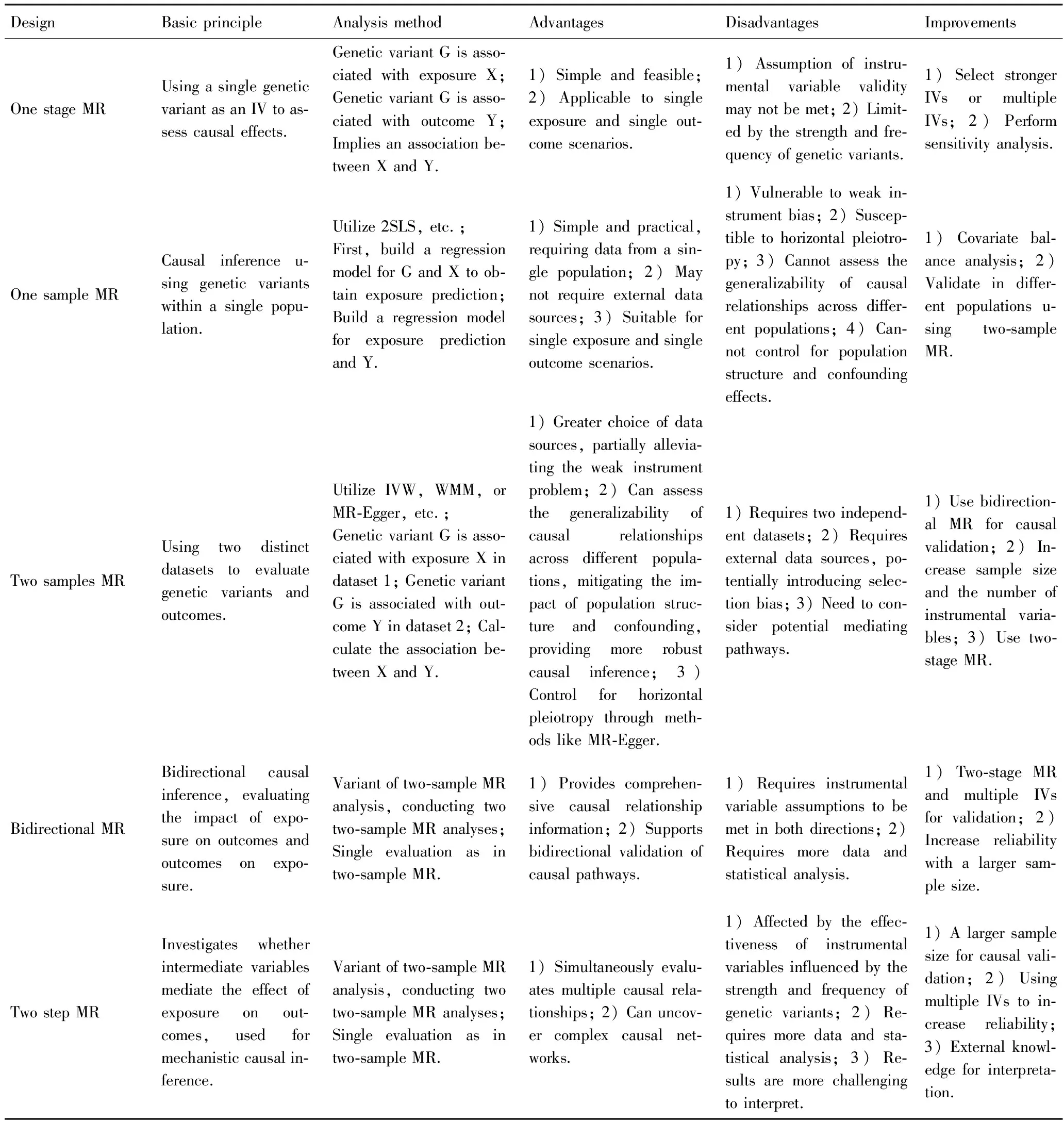
表1 常见的孟德尔随机化设计类型特点Tab.1 Common designs of Mendelian randomization designs and their characteristics
2 MR分析的具体实施方案中的关键挑战及优化
2.1 如何选择可满足MR分析的核心假设的IV?
本文第二部分提到过MR分析的核心假设有3个:相关性、独立性和排他性。
MR分析的首要步骤是选择满足MR分析的核心假设的IV。在实际研究中,如果关联的暴露是某个mRNA表达水平或循环中的某种蛋白质时,一般用单个基因的变异;而当关联的暴露是某个复杂性状,也就是遗传学概念中的多基因性状时,则IVs需要用多个基因的区域表示。确保IV与目标表型特征X之间存在稳健的强关联是确保因果推断可靠性的首要步骤,由于GWAS的显著性阈值设定是根据同时检测百万变异进行显著性矫正,即5×10-8,因此,在IV的筛选中一般以与暴露关联P<5×10-8作为显著性阈值。事实上,在3个核心假设中满足相关性是最容易实现的,而确保满足独立性和排他性需要注意以下几个关键点:
1)破坏相关性和独立性原则的“弱工具偏倚”
近年来GWAS和QTL作为复杂性状关联研究的热点,研究量井喷,这些数据集的公开共享极大的扩充了研究人员对IV的选择余地,但根据“常见疾病,常见变异”思想,即便达到显著性阈值,其实筛选出的每个SNP对表型的贡献都是微效的,不仅如此,对于复杂性状来说,整体遗传对于暴露的解释都仅为较小的一部分,使得MR研究比传统的流行病学估计具有更宽的置信区间(confidence interval,CI)[21];此外,受到混杂因素影响,即遗传变异通过暴露外的其他因素影响结果,因此虽然IV技术在有混杂因素的情况下是近似无偏倚,但IV在有限的样本量下,会出现遗传变异只能解释小部分暴露或关联强度不够的情况,被称为“弱工具偏倚”[12-13]。
弱工具偏倚的大小取决于遗传变异与暴露之间的关联强度,单样本MR研究中弱工具偏倚会偏向产生假阳性的结果,而两样本MR研究中会倾向低估关联的结果[22]。在研究中,须通过F统计量(F-statistic)来衡量、评估及排除。具体来说,在回归模型中,1/F统计量为回归方程中IV估计量的偏差与观察估计量的比值,如IV估计量的偏差是观察估计量偏差的10%时,F=10。F统计量的值越大,说明模型中的IV对暴露X的解释能力越强,因此,将F>10作为经验阈值判定IV的强度。R2及统计功率等也被用于剔除弱工具变量。
2)违反独立性原则的连锁不平衡和人口分层
在某些基因座位上,变异位点可能在进化过程中保持在一起,这种现象被称为连锁不平衡(linkage disequilibrium)。当两个或多个位点之间存在连锁不平衡时,意味着一个位点上的变异信息可能提供了有关另一个位点的信息,在作为IV候选时,可能违反独立性原则。另一个需要关注的则是人口分层(population stratification),指研究受试者群体由于地理、种族和族群差异,分为不同的亚群或亚型,这些亚群之间可能存在遗传和/或环境上的差异。这些差异可能导致了不同亚群之间基因型、表型和关联结构的差异,干扰特征相关的遗传因素的筛选。
为了避免这样的影响,在MR实践当中,可通过使用多个独立基因位点作为IV的组合,减轻单个IV的连锁不平衡的影响,同时使用协变量(例如,如通过主成分分析获得人口学特征)进行协变量平衡检测(covariate balance testing),检测不同人口子集之间的差异,并采取合适的统计方法来校正差异,确保MR分析中独立性原则的满足,减轻连锁不平衡和人口分层对研究结果的潜在干扰,提高因果推断的可靠性。
3)违反独立性和排他性原则的“水平多效性”
敏感性分析是评估结果的关键一步,虽然这步操作通常在流程中置于因果估计之后,但它的本质是用于检验因果估计对基础假设的依赖程度,有助于评估结果的可靠性,包括:①异质性检验(heterogeneity test): 用于评估在不同基因位点上的效应估计值之间是否存在显著的异质性(变异)。如果不同基因位点的效应估计值之间存在显著的异质性,那么可能存在基因位点间的不一致性,这可能影响到因果估计的稳健性。②水平多效性检测(horizontal pleiotropy detection):指一个遗传变异对多个相关性状(包括暴露因素X和结果Y)产生影响,而不仅仅影响研究关注的因果路径。如果存在水平多效性,那么IV可能不满足MR的假设,从而导致因果估计的偏差。③逐个剔除检验(leave-one-out test):通过逐步排除每个基因位点,重新进行因果估计,以评估每个基因位点对结果估计的影响。这有助于确定某个特定基因位点是否对因果估计产生重大影响,以及它是否主导了因果关系的估计。
2.2 因果估计的统计学方法如何选择?
一旦IV的有效性得到确认,就可以使用它们进行因果估计。在不同的MR设计类型中的因果估计方法主要有:
1)2SLS[23]:选择一个或多个IV,与暴露X(如表型)相关,但与结果Y的因果关系较弱。利用所选的IV与X之间的关系,进行第一阶段回归分析,IV与X的线性回归模型,计算IV对X的影响,获得暴露因素预测值(predicted value,P)。第二阶段使用P作为因变量,对Y进行回归分析,估计IV对Y的影响,从而间接估计X对Y的因果效应。
2)逆方差加权法(inverse variance weighted,IVW)[14]: 这是MR研究中最常用的方法之一。IVW方法将每个基因位点的效应估计值按照其方差的倒数进行加权平均,得到最终的因果估计值。这种方法假设基因位点的效应估计值是无偏的,并且没有遗传变异间的相互作用。
3)加权中位数法(weighted median method,WMM)[24]:与IVW方法不同,加权中位数法并不要求所有基因位点的效应估计值都是无偏的。它选择中位数效应估计值作为因果估计的点估计,通过将基因位点的效应估计值和方差进行加权。这种方法在一些情况下对异常值更具鲁棒性,因为它不受单个基因位点的影响。
4)MR-Egger法[25]:MR-Egger方法是用于处理IVW方法中可能存在的拮抗性或放大性偏差的一种技术。它与IVW最大的区别是在回归模型中保留“截距”,允许基因位点的效应估计值存在某种程度的偏斜,可以检测和校正因拮抗性或放大性而引起的估计偏差。MR-Egger法通过拟合一个带有截距的回归模型,估计因果效应并进行偏斜校正。
那么众多算法的分析结果中如何取舍呢?2SLS常用于对单样本MR进行分析,而在两样本MR及其变体的分析中可遵循:①在没有异质性和多效性的情况下,首选使用IVW方法的估计结果;②如果存在异质性但没有多效性,首选使用WMM方法的结果,也可以考虑使用IVW的随机效应模型;③当存在多效性时,首选使用MR-Egger方法计算出的结果。
此外,研究人员还通过更新算法模型来提升结果的可靠性,如新模型MRMix通过放宽IV与X关联的阈值纳入了更多的IV进行研究,同时通过采用正态混合模型来描述潜在效应大小分布,提供了几乎无偏或更小偏差的因果效应估计,提高了效能的同时也提高了精度。采用该方法的研究[26]显示HDL-C和TG对CAD风险没有因果效应。
2.3 结果的外推及如何进行生物学解读?
当研究人员获得MR分析的结果后,如何进行结果的外推及生物学解读仍须谨慎,在解读的过程中可能涉及到以下几个问题:
1)时间尺度和发展补偿:MR研究涉及的遗传变异是终生变异的结果,而病理水平的发展是逐渐累积并不可逆的,且生物体在不同时空有不同的遗传效应;
2)常规水平与病理水平:在使用MR研究分析结果作为药物筛选的理论支持时,需要考虑MR研究通常关注常规的暴露水平,而难以评估病理水平的短期靶向干预效果;
3)小差异的外推:由于遗传变异引起的暴露变化通常很小,而病理变化或药物干预的效果可能更加剧烈,所以需要更大的样本量进行MR分析来检测效应,且需要依赖外推法来估计干预效果;
4)遗传和干预效应的不同途径:遗传变异和干预措施通常不会对暴露产生相同的影响机制,不同途径和交互效应可能会导致不同的结果。
因此,在生物学外推和解读时需要谨慎考虑统计学和生物学的复杂性,以确保结果的生物学解释具有可信度。
2.4 其他需要考虑的问题
除了以上几点问题外,还有一些问题值得考虑。由于绝大多数GWAS研究主要关注人类基因组,因此在基于GWAS结果的MR研究中,IV的选择具有一定局限性,即只考虑了宿主基因组。那么海量的宏基因组数据可否提供IV帮助人们更好地了解疾病的全貌?刚刚提到的时间尺度和发育问题,最直观的就是发育和衰老研究中的表观遗传学范畴是否可以进行MR研究?大多数的疾病都是基因-环境共同影响,那么如何将基因和环境因素共同融入MR研究?这就催生了基于多组学汇总数据的MR分析方法来确定疾病中相关的假定因果效应和潜在机制,以更全面地理解疾病和健康的复杂性。如:宏基因组研究关注微生物群落、细菌、病毒等宿主以外的基因组,研究人员可以探索通过宏基因组测序获得的mbQTL数据进行MR研究,建立mbQTLs与健康、疾病和代谢相关的潜在关系[27];如果研究人员更关注基因表达、DNA甲基化、组蛋白修饰等表观遗传学变化对疾病和生理过程的影响,可利用mQTLs数据寻找合适的IV,进行MR分析,确定表观遗传学变化与特定疾病或生理现象的因果关联[28];群体规模足够大的研究可以用于执行基因-环境MR,以确定特定基因与环境因素如饮食、生活方式、药物暴露等之间的互动,从而更好地理解这些相互作用对健康和疾病风险的影响[29]。
3 MR的应用前景
MR作为“大自然馈赠的RCT”,其应用前景在生物医学研究和临床实践中具有重要的意义。尽管MR并不能完全替代RCT,但它提供了一种有用的补充手段,尤其适用于以下情况:①探索因果关系:MR可以帮助研究人员确定可改变的风险因素与结果之间的因果关系。通过利用自然界中存在的遗传变异,MR可以模拟随机对照试验的特性,从而提供更接近因果关系的结果[30]。②选择治疗干预靶点:对于临床干预的目标选择,MR提供了有价值的流行病学方法。通过评估某个生物标志物或治疗目标与特定疾病之间的因果关系,研究人员可以优先选择有效的干预靶点,从而提高治疗效果[31-32]。③长期基于人群的干预:MR估计尤其适用于长期基于人群的干预研究。在这种情况下,难以实施大规模的随机对照试验,而MR可以提供一种更可行的选择。
然而,需要注意的是,虽然MR可以为临床干预的效果方向提供定性信息,但遗传推导的估计可能与实际干预效果大小不一致。因此,在使用MR方法时,需要将估计结果与实际临床数据相结合,进行综合评估。综合来看,MR方法在探索因果关系、选择治疗干预靶点等方面具有广阔的应用前景。随着基因组学和遗传研究的不断发展,MR将继续为生物医学研究和临床实践提供有力的支持和指导。然而,在使用MR方法时,需要谨慎选择工具变量,并结合实际临床数据进行综合评估,以确保结果的准确性和可靠性。
致谢:本文的整体思路由丁卫教授指导完成,特此感谢!
利益冲突所有作者均声明不存在利益冲突。
作者贡献声明王晶:论文撰写;张国燕:论文修改;程杉:命题的提出、设计。
