干酪乳杆菌(Lactobacillus casei)HDS-01甘露聚糖酶基因异源表达及功能验证
2023-12-18杨若兮张鑫赵丹
杨若兮,张鑫,赵丹,3
(1黑龙江大学生命科学学院/农业微生物技术教育部工程研究中心/黑龙江省寒区植物基因与生物发酵重点实验室/黑龙江省普通高校微生物重点实验室,哈尔滨 150080;2长春金赛药业有限责任公司,长春 130012;3河北省农业生态安全重点实验室,河北秦皇岛 066102)
0 引言
甘露聚糖是一种结构复杂的生物聚合物,由于粘度高等特点,难以被人体直接消化利用。β-甘露聚糖酶(EC 3.2.1.78,β-mannanases,以下简称甘露聚糖酶)可以从甘露聚糖分子主链的内部随机水解β-1,4 糖苷键[1],降解甘露聚糖。魔芋粉的主要成分是魔芋葡甘露聚糖(Konjac glucomannan,KGM),由甘露糖和葡萄糖以β-1,4-糖苷键键合而成,是一种优质的膳食纤维[2]。KGM作为一种潜在的益生元,具有降低体重和预防糖尿病等生理功效,在食品工业中得到广泛应用[3]。魔芋葡甘低聚糖(Konjac oligo-glucomannan,KOGM)是降解KGM 形成的功能性低聚糖,作为一种重要的益生元,由于其原料易得、成本低廉等特点,具有良好的发展前景[4]。
微生物来源的酶具有产酶活性高、生产成本低等优势。常见产酶真菌有曲霉属(Aspergillus)、青霉属(Penicillium)及木霉属(Trichoderma)等[5-6]。与真菌相比,细菌发酵周期短,大多分泌胞外甘露聚糖酶,稳定性更高[7-8],使酶的分离纯化更容易[9]。大肠杆菌(Escherichiacoli)的遗传背景清晰,同时具有培养简单、生长迅速等特点,是优良的原核表达宿主[10]。至今已有不同来源的甘露聚糖酶基因被成功地从微生物中克隆并在异源表达菌株中获得了重组酶[11-12]。张丹阳等[13]从枯草芽孢杆菌(Bacillussubtilis)G1 中克隆出β-甘露聚糖酶基因BsmanA,将该基因与表达载体pACYCDuet-1 连接并转化到E.coliBL21(DE3)中,实现β-甘露聚糖酶基因的异源表达。目前研究较多的微生物有芽孢杆菌属(Bacillus)等,但此类微生物安全性较低。
乳酸菌(lactic acid bacteria,LAB)作为公认的安全(generally recognized as safe,GRAS)菌株[14],它直接分泌的胞外甘露聚糖酶具有安全保障[5,15-16]。目前已报道的直接产LAB 甘露聚糖酶菌株有干酪乳杆菌(Lactobacilluscasei)HDS-01、乳酸片球菌(Pediococcus acidilactici)M17、植物乳杆菌(Lactiplantibacillus plantarum)M24、绿色魏斯氏菌(Weissellaviridescens)LB37 和植物乳杆菌(L.plantarum)ATCC®14917TM[17]。天然LAB 产甘露聚糖酶存在产量较低,难以满足工业化应用需求。乳酸菌作为革兰氏阳性细菌,转化困难,遗传改造难度大,因此现有研究集中在这些菌株甘露聚糖酶蛋白的纯化、性质和应用,尚未涉及蛋白编码基因。
本研究以课题组前期分离自酸菜发酵液的一株产甘露聚糖酶的L.caseiHDS-01 为出发菌株[18],该菌株已完成全基因组测序(登录号:CP001084.2)。将L.caseiHDS-01 全基因测序结果与NCBI 数据库中已知甘露聚糖酶氨基酸序列进行本地BLAST比对,获得序列相似性较高的基因,基因组中编号为2884(以下称为基因M1),预测功能为甘露糖苷酶。甘露糖苷酶属于糖苷水解酶家族,推测有甘露聚糖酶活性。本研究首次将LAB甘露聚糖酶基因异源表达,以重组菌株的产酶效果和重组酶对KGM的降解效果验证甘露聚糖酶基因的功能。研究结果为乳酸菌甘露聚糖酶基因的遗传修饰和甘露聚糖酶蛋白的定向改造提供依据,也拓展了人们对乳酸菌聚糖代谢能力的认识。
1 材料与方法
1.1 试验材料
1.1.1 供试菌株和质粒L.caseiHDS-01,分离自酸菜发酵液,作为出发菌株;E.coliDH5α,购于上海唯地生物技术,用于克隆转化;E.coliBL21(DE3),购于上海唯地生物技术,作为重组蛋白表达的宿主。质粒pET-28a,长度为5369 bp,有卡那霉素抗性基因(Kanr),T7启动子,T7 终止子,6×His 标签,受IPTG 调控表达,由本实验室保存,在试验中作为重组蛋白表达载体;pET28a-M1,长度8051 bp,有卡那霉素抗性(Kanr),带有M1基因片段(2640 bp),由本实验室自行构建,用于表达重组蛋白M1。
1.1.2 培养基
(1)MRS培养基:牛肉膏1 g,蛋白胨1 g,葡萄糖2 g,酵母提取物0.5 g,磷酸氢二钾0.2 g,柠檬酸铵0.2 g,无水亚硫酸钠0.01 g,硫酸镁0.02 g,无水乙酸钠0.5 g,硫酸锰0.005 g,吐温-80 0.1 mL,蒸馏水0.1 L;pH 5.5,于121℃条件下灭菌15 min。
(2)LB培养基:蛋白胨0.1 g,酵母提取物0.5 g,氯化钠0.1 g,蒸馏水0.1 L;pH 7.0,于121℃条件下灭菌15 min。
(3)魔芋粉产酶培养基(KGM):魔芋粉0.6 g,牛肉膏0.1 g,蛋白胨0.1 g,葡萄糖0.1 g,酵母浸粉0.5 g,磷酸氢二钾0.2 g,柠檬酸铵0.2 g,无水亚硫酸钠0.01 g,硫酸镁0.02 g,无水乙酸钠0.5 g,硫酸锰0.005 g,吐温-80 0.1 mL,蒸馏水0.1 L;pH 5.5,121℃灭菌15 min。
1.2 基因M1异源表达菌株的构建
1.2.1 基因M1片段的获取和重组质粒pET28a-M1的构建根据L.caseiHDS-01基因组测序结果中的基因M1序列及表达载体pET-28a设计引物如下:M1-up,序列(5’→3’)为gtgccgcgcggcagccatatg ATGCAGAAA GTTCATGTTA TTGCC;M1-down,序列(5’→3’)为accagtcatgctagccatat gTCATAACGACTCCTTTTTCGATGC。
PCR 反应体系:Mix (green) 45 μL、基因组DNA 1 μL、M1-up 2 μL、M1-down 2 μL,经98℃预变性2 min,后经98℃变性10 s、60℃退火15 s、72℃延伸1 min,循环35次,最后72℃终延伸5 min。回收目的片段。利用限制性内切酶NdeI 对表达质粒pET-28a 进行单酶切,以获得与DNA片段含有15~20 bp互补末端的线性化载体。
酶切体系:限制性内切酶NdeI 1 μL、质粒pET-28a 1 μL、Cutsmart buffer 5 μL,37℃反应30 min。琼脂糖凝胶回收试剂盒纯化线性化载体,将线性化载体做数个梯度稀释,原液和稀释后产物各取1 μL上样进行电泳,与标准的DNA 定量Marker 比较条带亮度以确定其近似浓度。
线性化载体和纯化后M1片段按比例混合后在重组酶Exnase II 催化下实现两线性化DNA 的体外环化。采用热激法将重组反应体系转化E.coliDH5α感受态细胞。以添加终浓度为100µg/mLKan的LB 固体培养基筛选阳性重组克隆。以筛选菌落提取的质粒为模板,利用T7-up 和T7-down 为引物进行PCR 验证及酶切验证目的基因片段是否正确整合到表达载体上。引物设计如下:
T7-up,序列(5’→3’)为CGATCCCGCGAAATTA ATACGACTC;
T7- down,序列(5’→3’) 为 TCAGCTTCCT TTCGGGCTTTGTTAG。
将含有阳性质粒的菌株命名为E.coliDH5αpET28a-M1。
PCR 反应体系:T3 Super PCR Mix 22 μL、Plasmid 1 μL、T7-up1 μL、T7-down 1 μL。
酶切体系:限制性内切酶SacI 1 μL、Plasmid DNA 1 μL、Cutsmart buffer 5 μL,37℃反应30 min。
1.2.2 重组质粒pET28a-M1转化E.coliBL21(DE3) 用热激法将重组反应体系转化至E.coliBL21(DE3)感受态细胞中,以含有Kan浓度为100µg/mL的LB平板中挑取重组子,T7-up和T7-down为引物进行PCR验证。
将阳性克隆至接种至LB液体培养基(含Kan终浓度为100 µg/mL)中,37℃、150 r/min,摇床培养过夜。使用质粒小提试剂盒提质粒进行PCR 验证及酶切验证目的基因片段是否正确整合到表达载体上。将验证后的阳性克隆命名为E.coliBL21(DE3)-pET28a-M1。
PCR 反应体系和酶切体系同E.coliDH5αpET28a-M1。
1.2.3 IPTG诱导重组蛋白M1的表达从阳性菌落平板中挑取单菌落接入LB液体培养基中,加入Kan至终浓度为100 µg/mL,37℃、150 r/min,摇床培养过夜。按1%接种量接入到含有250 mL LB液体培养基中,加入Kan至终浓度为100µg/mL,37℃、150 r/min,培养2~3 h至OD600为0.6~0.8之间。以0号管为对照组,在其余管中加入150 μL 的IPTG(终浓度50 mmol/L),诱导表达。从每组试管中取0.5 mL 菌液加入到1.5 mL 离心管中,6000 r/min,5 min 去上清,留沉淀。在每支离心管中加入60 μL的1×PBS和15 μL的5×Loading Buffer悬浮沉淀。100℃煮样10 min。取出后离心6000 r/min,5 min,SDS-PAGE检测蛋白表达情况。
1.3 重组蛋白M1的纯化
20 mmol/L PB(pH7.2),300 mmol/L NaCl 含1%Triton X-100,2 mmol/L EDTA,5 mmol/L DTT 的缓冲液洗涤后,使用20 mmol/L PB(Ph 7.2),300 mmol/L NaCl,8 M Urea,20 mmol/L Imidazole缓冲液溶解包涵体同时平衡Ni-IDA柱,上样后用不同浓度咪唑的平衡缓冲液洗脱目标蛋白。
1.4 重组菌株蛋白M1酶活力的测定
以未转化质粒的空菌株为对照,检测重组菌株的发酵液上清、菌体破壁后上清、包涵体溶解后上清以及蛋白复性后的上清的甘露聚糖酶活力。
按照公式(1)计算甘露聚糖酶活力[19]。
1.5 重组菌株蛋白M1蛋白含量的测定
采用Bradford法进行蛋白质含量的测定[20]。
1.6 高效液相色谱测定酶解糖类物质聚合度及含量
将Ni 柱亲和层析纯化后的重组蛋白M1 置于0.6%魔芋粉的PBS 磷酸缓冲液中,50℃条件下分别反应1、2、3、4、5、6 h。
1.7 数据分析及处理
本研究数据由3个独立样本的平均值及其标准差显示。绘图软件为Origin 2018和SigmaPlot 10.0。
2 结果与分析
2.1 重组质粒pET28a-M1的构建
PCR 扩增结果如图1(A)所示,在2600 bp 左右处有单一明亮片段,与预期大小相符。
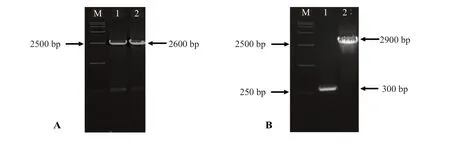
图1 L.casei HDS-01M1基因PCR结果(A)和阳性克隆子质粒PCR验证结果(B)
使用限制性内切酶NdeI 对表达质粒pET-28a 进行单酶切,获得线性化载体,在5400 bp左右处有单一明亮条带,与预期大小符合。将线性化的pET-28a 载体和M1片段按比例混合后,在重组酶Exnase II 催化下,线性化载体与DNA片段完成体外环化。重组质粒M1的PCR验证结果如下:泳道1~4均出现了阳性目的条带,大小约为2900 bp。结果表明,M1片段成功整合到了表达载体pET-28a上。
使用质粒小提试剂盒提取上述阳性菌株中的重组质粒,酶切验证及PCR 验证的结果如下:筛选得到的M1阳性菌株质粒的条带大小约为7900 bp,包含了目的基因M1片段序列,与预期大小相符。
2.2 重组质粒pET28a-M1转化E.coli BL21(DE3)
将重组质粒pET28a-M1转化至E.coliBL21(DE3),单菌落PCR 结果如图1(B)所示。图1(B)中泳道1 为空质粒pET-28a PCR 的验证结果,约为300 bp,泳道2 为重组质粒pET28a-M1PCR 验证结果,约为2900 bp,与预期大小相符。
2.3 IPTG诱导M1表达与SDS-PAGE电泳分析
图2(A)为使用IPTG 诱导重组质粒表达后的蛋白表达结果。泳道1 和3 分别为对照菌株E.coliBL21(DE3)发酵液和发酵液上清破壁缓冲液的蛋白表达情况。泳道2 和4 分别为对照菌株E.coliBL21(DE3)-pET28a-M1发酵液和发酵液上清破壁缓冲液的蛋白表达。泳道2在100 kDa左右出现了清晰的条带,但与泳道4却没有,这说明可能是因为在菌体破碎过程中,重组蛋白M1形成了不溶性的包涵体蛋白。
如图2(B)所示,重组蛋白M1经8 M Urea溶解后,泳道2与对照菌株E.coliBL21(DE3)(泳道1)相比,在100 kDa 左右处出现了清晰的条带,蛋白分子量与理论值相一致,与图2(A)结果共同说明重组蛋白M1 以包涵体的形式表达。
2.4 重组蛋白纯化及体外功能验证
2.4.1 纯化M1 重组蛋白E.coliBL21(DE3)-pET28a-M1菌株经IPTG诱导、PBS缓冲液重悬菌液、超声条件下裂解细胞、按包涵体的纯化方式纯化重组蛋白M1后,收集各个阶段组分进行SDS-PAGE 分析检测(图3A)。泳道6 为未诱导条件下细胞破碎后的沉淀。泳道7为诱导条件下细胞破碎后的沉淀。泳道7与阴性对照(泳道1)相比在100 kDa附近有特异性条带,说明M1在细胞破碎后形成包涵体。泳道8为复性后经Ni-IDA Resin 树脂亲和层析纯化后的蛋白,条带单一,说明重组蛋白M1纯化成功。
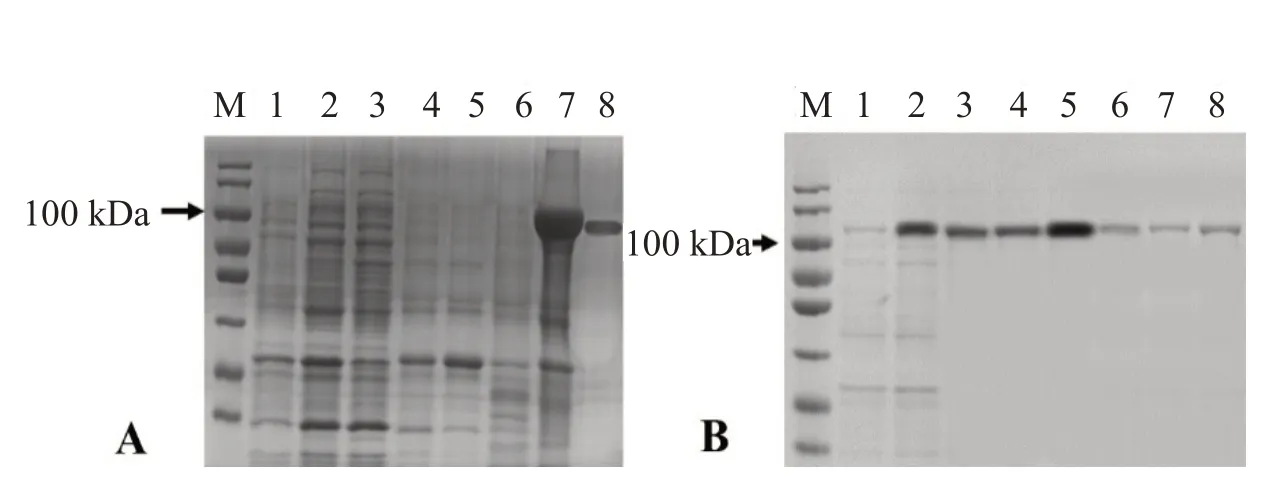
图3 SDS-PAGE分析M1蛋白纯化结果(A)和不同浓度咪唑纯化M1重组蛋白(B)
用含有不同咪唑浓度的洗脱液洗脱目的蛋白M1,结果如图3(B)所示。不同浓度的洗脱液都可以将目的蛋白M1 洗脱下来,但效果有所差异,其中100 mmol/L 浓度咪唑的洗脱液纯化的蛋白条带颜色最深,效果最好。
2.4.2 重组蛋白M1 酶活力测定如图4 所示,E.coliBL21(DE3)的发酵在发酵液上清中检测出的酶活较高。E.coliBL21(DE3)-pET28a-M1在离心收集菌体、PBS 缓冲液重悬菌液、超声裂解细胞、离心收集上清时,酶活达到最大,为31.17±0.55 U/mL。
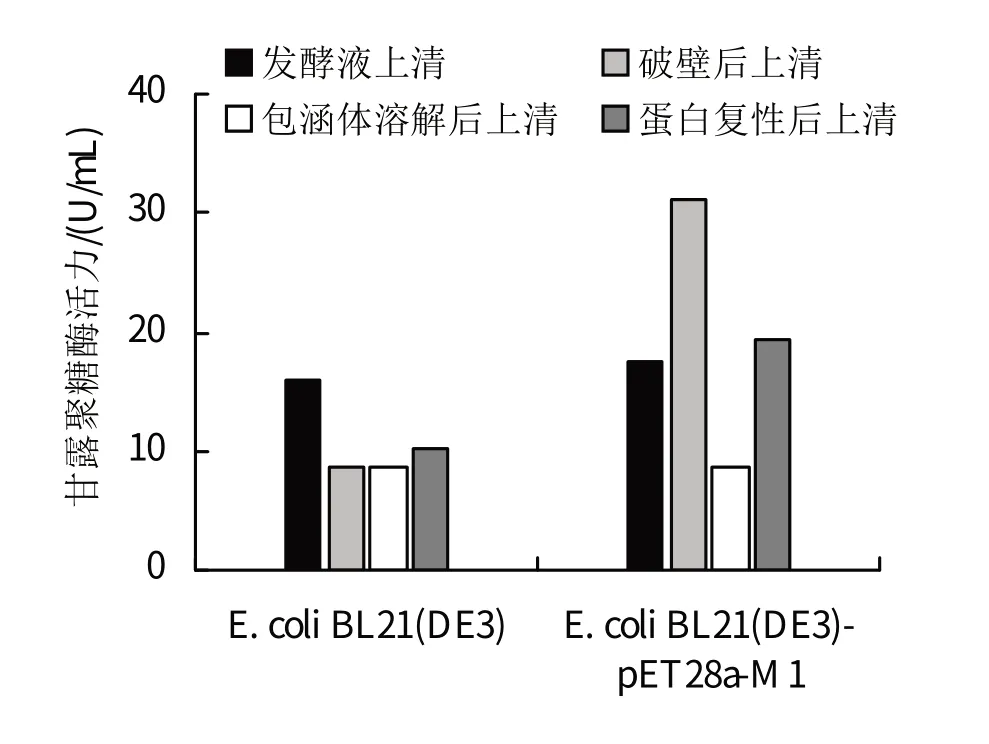
图4 基因工程菌株甘露聚糖酶活力测定
2.4.3 重组蛋白M1 蛋白浓度检测图5 为相同条件下的重组菌株蛋白浓度变化情况。E.coliBL21(DE3)的蛋白浓度经不同处理后,蛋白浓度逐渐降低,表示在整个处理阶段,蛋白损失较高。而E.coliBL21(DE3)-pET28a-M1包涵体溶解后上清的蛋白质浓度和发酵液上清的蛋白质浓度相当,说明重组蛋白M1 大部分以包涵体形式存在。在复性过程中,重组蛋白M1 的蛋白浓度也出现损失,同时酶活性降低。
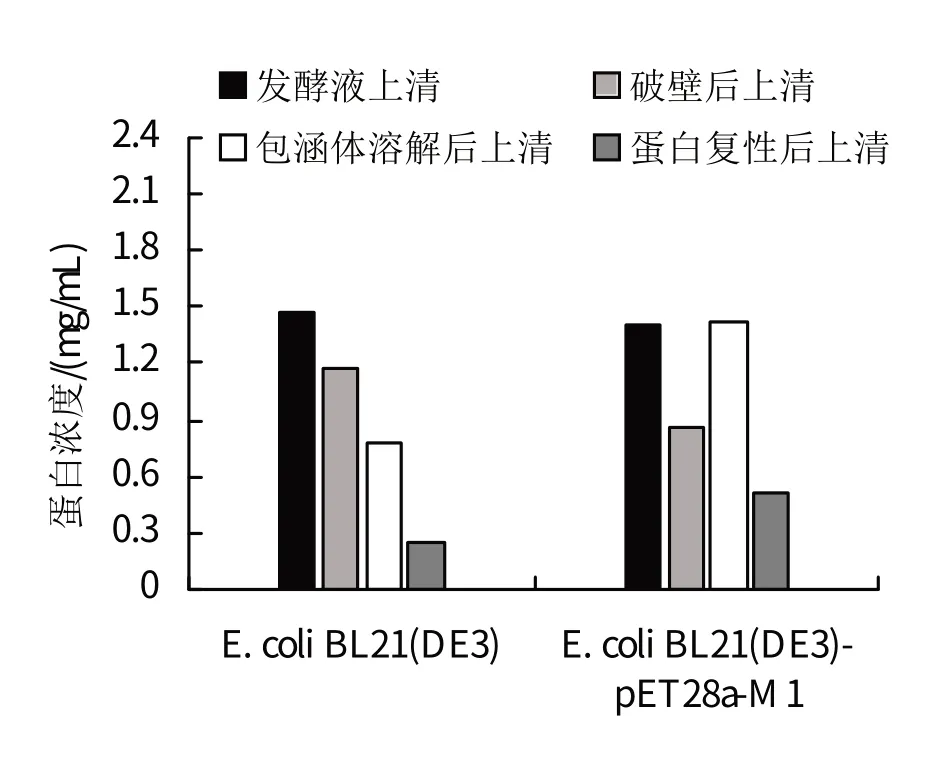
图5 基因工程菌株蛋白浓度测定
2.4.4 糖类物质的变化情况重组蛋白M1降解魔芋甘露聚糖的产物有甘露糖、甘露二糖、甘露三糖和甘露四糖,结果如图6 所示。在反应前3 h 中,甘露三糖和甘露四糖的含量明显高于甘露糖和甘露二糖,证明此时重组蛋白M1降解多聚糖生成三糖和四糖。3 h后,甘露三糖和甘露四糖浓度逐渐降低,甘露糖和甘露二糖浓度逐渐升高。
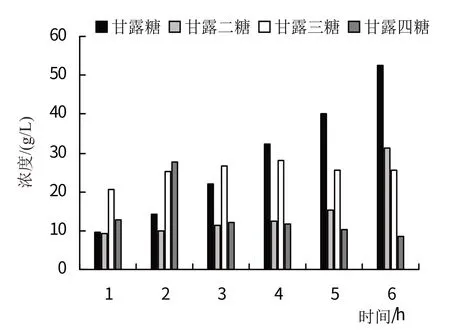
图6 重组蛋白M1水解魔芋粉甘露低聚糖浓度变化
多重比较结果显示,在反应前3 h,甘露三糖或四糖的浓度显著高,3 h 后,甘露糖的浓度显著高。随着反应的进行,甘露糖和甘露二糖浓度呈逐渐上升,甘露三糖和甘露四糖浓度趋势呈现先上升后下降,分别在4 h 和2 h 累计浓度达到最高,为27.85±0.03 g/L 和28.17±0.19 g/L。这是由于2 h后甘露三糖在重组酶的降解作用下,被降解为了单糖和二糖,而甘露四糖被降解为了单糖、二糖和三糖。与甘露四糖相比,三糖的变化速度较稳定,可能是由于甘露三糖的生成速率和消耗速率差异较小。
3 讨论与结论
利用原核表达载体诱导表达可能导致蛋白合成速度过快,造成错误折叠和异常聚积,形成无生物活性、不可溶的包涵体。在本研究中,重组蛋白M1 在表达过程中以包涵体蛋白形式存在,经8 M Urea 溶解、亲和层析纯化和透析复性最终得到了大小为98.72 kDa重组蛋白。马鑫等[21]将Bacillussp.中的甘露聚糖酶基因与表达载体pET-28a 连接,转化E.coliBL21,使用IPTG 作为诱导物,37℃培养3 h 表达重组甘露聚糖酶。但SDS-PAGE 分析表明,表达出的重组甘露聚糖酶主要以包涵体形式存在于细胞质中。朱泾等[22]利用PCR技术从硫磺矿硫化叶菌(Sulfolobussolfataricus)P2中扩增得到甘露聚糖酶基因,克隆至E.coli表达载体pET-30a 中构建重组质粒pET-30a-3007,并转化至E.coliRosetta。利用IPTG 诱导重组菌株pET-30a-3007,与本研究一致,SDS-PAGE检测得到的目的蛋白大部分以包涵体形式存在,分子量大小为70 kDa,与预测一致。
基因M1的预测功能为甘露糖苷酶,作为甘露聚糖酶的一种,甘露糖苷酶可以从非还原末端切割甘露聚糖主链[23]。ZHANG 等[24]将分离自小麦中的甘露糖苷酶基因(TaMan3A)异源表达在毕赤酵母(Pichia pastoris)中,重组基因可将半乳甘露聚糖和魔芋葡甘露聚糖的甘露聚糖聚合物水解为甘露二糖,甘露三糖和甘露糖,表明甘露糖苷酶具有直接将甘露聚糖降解为低聚糖的能力。使用重组酶水解甘露聚糖不仅能验证假定甘露聚糖酶的功能,也是制备甘露低聚糖较为常见的一种方法。者园园等[25]从魔芋基地筛选产甘露聚糖酶菌株并克隆甘露聚糖酶基因,通过电转化法至P.pastorisGS115 中实现异源表达重组酶水解魔芋粉的产物组分主要为甘露二糖和甘露三糖。甄红敏等[26]研究发现,与大部分GH26 家族β-甘露聚糖酶不同,PcMan26A 的最小水解底物为甘露五糖,能将甘露五糖水解为甘露糖和甘露四糖,不产生甘露二糖和甘露三糖。郭金玲等[27]利用薄层层析定性分析B.subtilis中甘露聚糖酶的降解魔芋粉的酶解产物主要为三糖及三糖以上的低聚糖。综上共同说明,来源不同甘露聚糖酶降解相同底物生成的产物和低聚糖含量存在差异。
本研究首次通过异源表达获得了乳酸菌来源的甘露聚糖酶的重组蛋白,SDS-PAGE 显示蛋白大小为98 kDa,复性后酶活和蛋白浓度分别为19.24±0.55 U/mL和0.51±0.01 mg/mL。以魔芋粉为底物验证了重组蛋白降解聚糖的功能,证实了乳酸菌基因组中甘露糖苷酶基因的编码蛋白具有甘露聚糖酶活性。研究成果有助于推进乳酸菌甘露聚糖酶的食品级工业化进程。
