Classification of patients with metastatic colorectal cancer into consensus molecular subtypes into real-world: A pilot study
2023-12-15JaimeGonzlezMonteroMauricioBurottoGuillermoValenzuelaDeboraMatelunaFlorenciaBuenAbadJessicaToroOlgaBarajasKatherineMarcelain
Jaime González-Montero,Mauricio Burotto,Guillermo Valenzuela,Debora Mateluna,Florencia Buen-Abad,Jessica Toro,Olga Barajas,Katherine Marcelain
Abstract BACKGROUND Colorectal cancer is a complex disease with high mortality rates.Over time,the treatment of metastatic colorectal cancer (mCRC) has gradually improved due to the development of modern chemotherapy and targeted therapy regimens.However,due to the inherent heterogeneity of this condition,identifying reliable predictive biomarkers for targeted therapies remains challenging.A recent promising classification system—the consensus molecular subtype (CMS) system—offers the potential to categorize mCRC patients based on their unique biological and molecular characteristics.Four distinct CMS categories have been defined: immune (CMS1),canonical (CMS2),metabolic (CMS3),and mesenchymal (CMS4).Nevertheless,there is currently no standardized protocol for accurately classifying patients into CMS categories.To address this challenge,reverse transcription polymerase chain reaction (RT-qPCR) and next-generation genomic sequencing (NGS) techniques may hold promise for precisely classifying mCRC patients into their CMSs.AIM To investigate if mCRC patients can be classified into CMS categories using a standardized molecular biology workflow.METHODS This observational study was conducted at the University of Chile Clinical Hospital and included patients with unresectable mCRC who were undergoing systemic treatment with chemotherapy and/or targeted therapy.Molecular biology techniques were employed to analyse primary tumour samples from these patients.RT-qPCR was utilized to assess the expression of genes associated with fibrosis (TGF-β and β-catenin) and cell growth pathways (c-MYC).NGS using a 25-gene panel (TumorSec) was performed to identify specific genomic mutations.The patients were then classified into one of the four CMS categories according to the clinical consensus of a Tumour Board.Informed consent was obtained from all the patients prior to their participation in this study.All techniques were conducted at University of Chile.RESULTS Twenty-six patients were studied with the techniques and then evaluated by the Tumour Board to determine the specific CMS.Among them,23% (n=6),19% (n=5),31% (n=8),and 19% (n=5) were classified as CMS1,CMS2,CMS3,and CMS4,respectively.Additionally,8% of patients (n=2) could not be classified into any of the four CMS categories.The median overall survival of the total sample was 28 mo,and for CMS1,CMS2,CMS3 and CMS4 it was 11,20,30 and 45 mo respectively,with no statistically significant differences between groups.CONCLUSION A molecular biology workflow and clinical consensus analysis can be used to accurately classify mCRC patients.This classification process,which divides patients into the four CMS categories,holds significant potential for improving research strategies and targeted therapies tailored to the specific characteristics of mCRC.
Key Words: Metastatic colorectal cancer;Targeted therapy;Consensus molecular subtypes;Personalized medicine
INTRODUCTION
Colorectal cancer (CRC) exhibits high incidence and mortality rates.At the time of diagnosis,approximately 25% of patients already present with metastatic disease,while 50% of those initially diagnosed with localized stages later develop disseminated disease[1].Recent years have seen significant advancements in systemic therapies for metastatic colorectal cancer (mCRC) patients,including diverse combination chemotherapy regimens,targeted therapy,immunotherapy,and multi-kinase inhibitors[2].Despite these improvements,patients’ responses remain variable and unpredictable due to the molecular heterogeneity of this disease.Thus,it is imperative to identify specific mutations for a personalized treatment approach[3].
Numerous efforts have attempted to identify distinct molecular mCRC phenotypes.In 2015,bioinformatic studies revealed a promising classification system with four consensus molecular subtypes (CMS)[4].This classification system has gained widespread clinical acceptance and is currently guiding various ongoing clinical trials[5].The four CMS are as follows: CMS1,or immune subtype,primarily affects young patients and exhibits rapid progression and resistance to conventional therapies.This subtype may benefit from aggressive chemotherapy and,potentially,immunotherapy.CMS2,or canonical subtype,is characterized by mutations in specific pathways linked to cellular metabolism.CMS3,or metabolic subtype,is characterized by mutations in pathways responsible for cellular metabolism,with a high prevalence ofKRASpathway mutations.Finally,CMS4,or mesenchymal subtype,is associated with mutations in fibrogenesis and epithelial-mesenchymal transition pathways,leading to a poor prognosis and a higher incidence of metastasis[5].To date,there is no established methodology for effectively classifying patients into CMS categories.However,given that each CMS is linked to distinct patterns of mutations and gene expression,it is plausible that a molecular biology workflow designed to identify specific mutations could help accurately classify patients into different CMS groups[6].Therefore,the objective of this study was to establish a workflow for assigning mCRC patients to CMS categories using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and next-generation sequencing (NGS) techniques.
MATERIALS AND METHODS
Study design and participants
In this observational study conducted between 2020 and 2023,we analyzed primary tumor tissue samples from mCRC patients who were receiving systemic treatment at the University of Chile Clinical Hospital.Colon or rectal tissue samples were collected through colonoscopy or surgical procedures.The samples were processed and stored according to protocols established by the Biobank of Tissues and Fluids at the University of Chile (http://biobanco.uchile.cl/).Both formalin-fixed paraffin-embedded (FFPE) tissue biopsies and fresh neoplastic tissue (frozen without fixation) were examined.
The inclusion criteria for this study were as follows: Patients diagnosed with unresectable mCRC (colon or rectal cancer) confirmed through histological diagnosis.Undergoing treatment at the University of Chile Clinical Hospital.Receiving systemic therapy in accordance with international clinical guidelines (National Comprehensive Cancer Network[7] and European Society of Medical Oncology)[8].Treatment regimens included chemotherapy (FOLFOX,CAPOX,or FOLFIRI) and targeted therapy (bevacizumab,aflibercept,cetuximab,panitumumab,regorafenib,and TAS102).Chemotherapy and targeted therapy regimens were selected by the physicians on a case-by-case basis.
The exclusion criteria for this study were as follows: Patients who underwent the removal of metastases (metastasectomy) before enrollment.Any comorbidity leading to a life expectancy of less than six months.Inability to maintain clinical follow-up.
RT-qPCR
The expression of TGF-β,β-catenin,and c-MYC was investigated as follows: RNA was extracted from FFPE tissue using the RecoverAllTMTotal Nucleic Acid Isolation Kit for FFPE (Invitrogen).Subsequently,the concentration of each RNA sample was determined using the Quant-iTTMRiboGreenTMRNA Reagent Kit (Invitrogen) on a Cytation 3 instrument (BioTek).RNA (1000) ng was then used to prepare cDNA with the AffinityScript qPCR cDNA Synthesis Kit (Agilent) according to the manufacturer’s instructions. Amplifications by qPCR (real-time PCR) was conducted in triplicate using the Brilliant II SYBR Green qPCR Master Mix kit (Agilent) on an Eco Real-time PCR System (Illumina).The following cycling conditions were used: an initial denaturation step at 95ºC for 10 min,then 40 cycles of amplification (each cycle is 10 s at 95ºC,30 s at 60ºC and 15s at 72°C).A melting curve for each primer ensured amplification of a single product.Finally,six FFPE non-tumour tissue samples treated in the same manner as the FFPE tumour tissues from each patient were included as controls.The relative expression was calculated using the ΔΔCt method[9] and normalized using expression levels of reference genes: B2M,PPIA,and RPLP0.Table 1 presents a summary of the primers used to conduct the RT-qPCR experiments[10-15].
NGS
The presence of genomic mutations was assessed using a 25-gene panel (TumorSec) as described by our team[16].The RecoverAllTMTotal Nucleic Acid Isolation Kit for FFPE was utilized to extract genomic deoxyribonucleic acid (DNA) from FFPE samples.Briefly,samples were incubated with 1 mL of Histo-Clear at 50°C for 3 min to remove paraffin.The supernatant was then removed,followed by two ethanol washes,and the residual ethanol was evaporated using a SpeedVAC (Thermo Scientific).The samples were then incubated overnight in a digestion solution containing proteases.The next day,the samples were incubated at 80°C for 15 min and an isolation additive was added and centrifuged.Subsequently,the supernatant was transferred to a filter column and centrifuged to isolate the RNA,which was then treated with DNase.The column contained the DNA,which was subsequently treated with RNase.The DNA and RNA were washed with wash buffers and eluted in elution buffer in separate tubes.
Quantification and quality analysis: The purity and quantity of DNA and RNA were determined by measuring absorbance at 260/280 nm with the PicoGreen assay (Quant-iTTMPicoGreen®dsDNA,Invitrogen) and the Quant-iTTMRiboGreenTMRNA Reagent Kit,respectively,on a Cytation 3 instrument (Biotek).Additionally,DNA quality analysis was conducted by measuring fragment size with the HS Genomic DNA Analysis Kit (DNF-488) (Agilent) on a Fragment Analyzer instrument (Agilent).As the extraction of genomic DNA from FFPE samples often results in low yields and degradation ranging from more than 1000 bp to less than 200 bp,fragments less than 200 bp were not used for library preparation due to excessive degradation.To ensure adequate DNA quantity,a minimum of four,6-μm FFPE sections per patient were used for sequencing.Moreover,each sample needed to contain more than 20% tumour content.
Library preparation: The KAPA HyperPlus Library Preparation Kit (Kapa Biosystems) was utilized to prepare DNA libraries.Library sizes and concentrations were verified for quality control purposes.The 260/280 nm ratio was measured with Cytation equipment and quantification was carried out using the Quant-iTTMPicoGreenTMdsDNA Assay Kit.Furthermore,library sizes were visualized using the HS NGS Analysis Kit in a Fragment Analyzer instrument.
NGS: NGS was conducted following a protocol previously published by our team[9].For sequencing,an equimolar pool of libraries (4 nM) was prepared,diluted,and denatured to achieve a final concentration of 9.4–9.5 pM according to guidelines in the "MiSeq System Denature and Dilute Libraries Guide" (Illumina).Paired-end sequencing (300 cycles) was performed using the Illumina MiSeq System (MiSeq Reagent Kits v2).Finally,bioinformatics analysis was conducted.
Classification of patients into CMS categories
Given the absence of a singular marker that differentiates each of the four CMS categories on its own,we developed a comprehensive protocol involving analysis by a Tumour Board consisting of experts in Molecular and Medical Oncology.Each case was individually assessed and the CMS was determined based on the criteria defined by Guinneyet al[4].The Tumour Board relied on patients’ clinical characteristics,mismatch repair (MMR) expression,and RT-qPCR and NGS results.Each patient’s CMS was determined by consensus among all committee members.Patients for whom a CMS consensus could not be reached were considered unclassifiable.
The Tumour Board employed the following criteria to classify each patient into one of the four CMS categories.It is important to note that none of these elements individually serve as a specific CMS marker;instead,classifications were based on the combination of multiple elements and reached through tumour board consensus.CMS1: presence ofBRAFmutation;MMR protein deficiency;low TGF-β,β-catenin,and c-MYC mRNA expression;and absence ofAPCorKRASmutations.CMS2 and CMS3: presence ofAPCmutation;absence ofBRAFmutation (with a predominance ofKRASmutations in CMS3);MMR-proficient;low TGF-β and β-catenin mRNA expression;and high c-MYC mRNA expression.CMS4: MMR-proficient;high expression of TGF-β and β-catenin mRNA;low expression of c-MYC mRNA;and presence of non-categorical mutations identified through NGS[6].
Ethics
All procedures conducted in this study were in full compliance with the ethical standards set by the Institutional and National Research Committee,as well as the principles outlined in the 1964 Declaration of Helsinki and its subsequent amendments.Ethical approval for this study was obtained from the Ethics Committee of the University of Chile Clinical Hospital and Faculty of Medicine prior to beginning the research.Informed written consent was obtained from all patients before their participation in the trial.
Statistics
Results are presented as the number and percentage of total patients included in this study.To determine the appropriate sample size,we considered the estimated prevalence of each mCRC CMS.According to previous work[4],the expected prevalence of each CMS is approximately 20%–25%.A sample size of 25 patients was deemed sufficient to analyze the prevalence and distribution of the different CMS categories.Indeed,prior research has utilized sample sizes of 20–30 patients;thus,a sample size of 25 patients is consistent with the literature.For the overall survival analysis of the studied patients,log-rank test was conducted using GraphPad Prism 10.0 software.
RESULTS
Between 2020 and 2023,a total of 26 patients with unresectable mCRC undergoing systemic treatment at the University of Chile Clinical Hospital were included in this study.Table 2 presents the demographic and clinical characteristics of the patients,including age,gender,primary tumour site,and the presence or absence of MMR proteins.Each patient is identified with a number from 1–26.
Molecular studies
Table 2 illustrates the results of an RT-qPCR-based gene expression analysis of TGF-β,β-catenin,and c-MYC in each of the patients studied.It is observed that the expression of these three genes is heterogeneous among patients.Table 3 provides a comprehensive overview of the mutations identified with the 25-gene TumorSec panel.The most frequently observedmutations were inKRAS,TP53andARID1A.All observed mutations were single nucleotide variants (SNVs) and two patients possessed deletions.
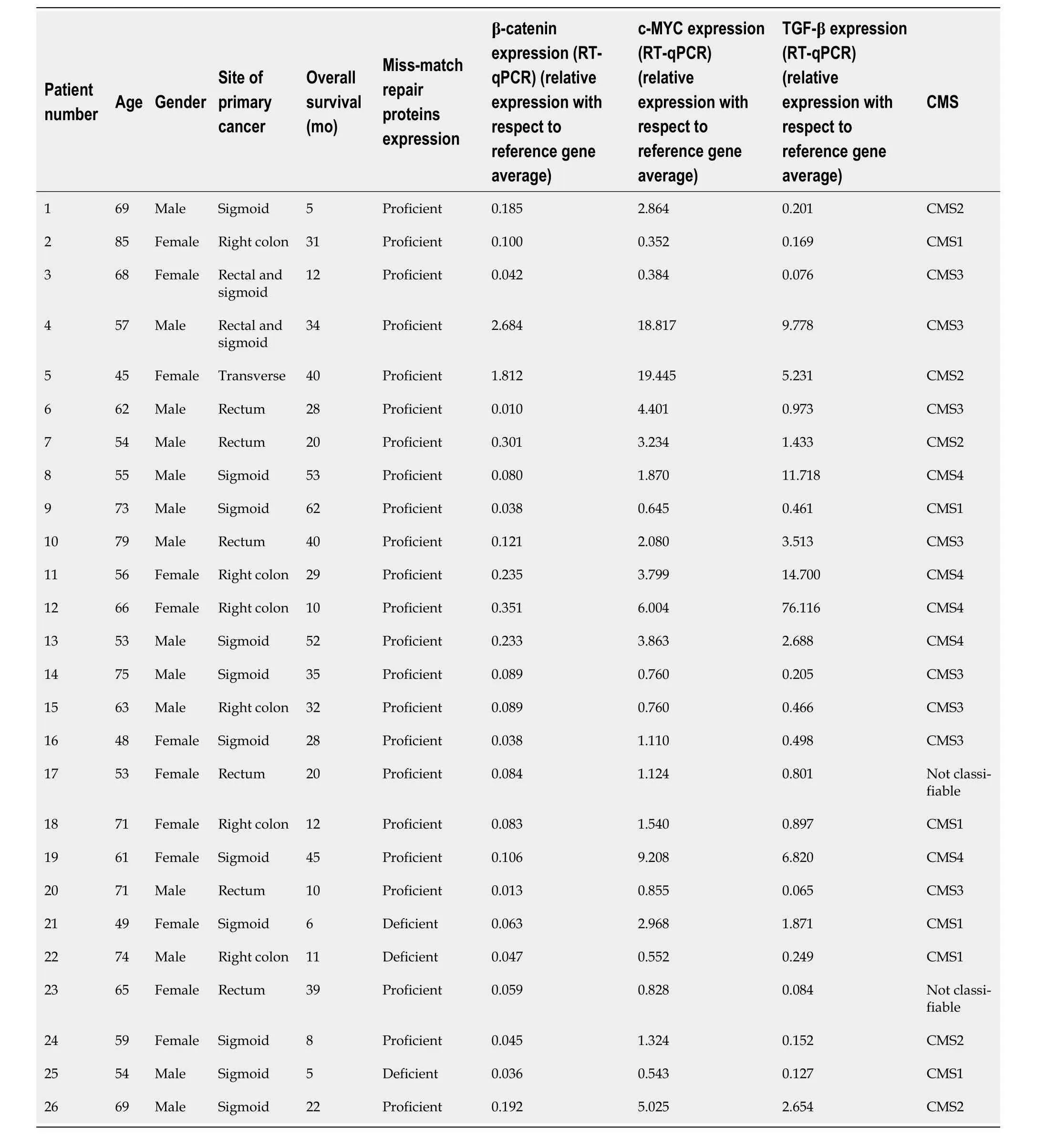
Table 2 Clinical characteristics,overall survival,and reverse transcription-quantitative polymerase chain reaction results of the n=26 patients included in the final analysis
Classification of patients into CMS categories
Out of the 26 patients analyzed,a specific CMS could be identified for 24 patients (92%) by clinical consensus by the Tumour Board.Two patients (8%) were found to be unclassifiable.Figure 1 illustrates the distribution of patients acrossthe four CMS categories.Specifically,23% (n=6),19% (n=5),31% (n=8),and 19% (n=5) were classified as CMS1,CMS2,CMS3 and CMS4,respectively.Remarkably,the percentage of patients classified into each CMS category closely aligns with findings reported by Guinneyet al[4].The median overall survival of the total sample was 28 mo (Figure 2A),and for CMS1,CMS2,CMS3 and CMS4 it was 11,20,30 and 45 mo respectively,with no statistically significant differences between groups (Figure 2B).
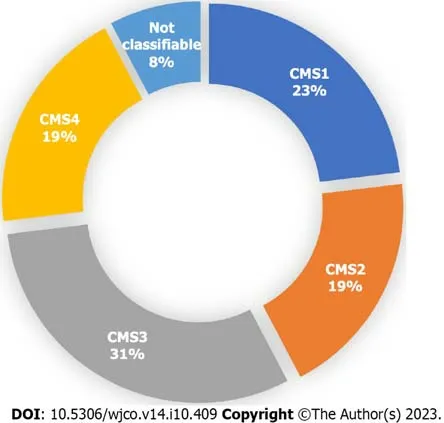
Figure 1 Proportion of patients in each consensus molecular subtype after analysis by the Tumour Board among the 26 patients included on the final analysis. A specific consensus molecular subtype (CMS) was successfully identified in 24 out of the 26 patients.CMS1 n=6.CMS2 n=5.CMS3 n=8.CMS4 n=5.Not classifiable n=2.Each patient underwent an individual assessment by the Tumour Board,and a consensus was reached to determine their molecular subtype.Classification was based on clinical and histological characteristics,as well as the results of RT-qPCR (β-catenin,c-MYC and TGF-β) and NGS (TumorSec panel).CMS: Consensus molecular subtypes.
DISCUSSION
The objective of the workflow outlined in this manuscript was to develop an RT-qPCR-and NGS-based method by which to classify mCRC patients into CMS categories.Our results demonstrate that it is possible to classify mCRC patients into a specific CMS in approximately 90% of the cases.

Figure 2 Classification of patients into consensus molecular subtype categories.A: Kaplan-Meier Curve with overall survival (OS) of the n=26 patients included on the final analysis.mOS=28 mo;B: Kaplan-Meier curve which shows OS of patients based on their molecular subtype classification.The median overall survival times were 11,20,30,and 45 mo for CMS1,CMS2,CMS3,and CMS4,respectively.There were no statistically significant differences observed among the studied groups (P=0.0968).
To date,there are no validated tools from prospective studies for classifying patients into the four CMS categories.Although genomic platforms such as ColotypeR[17] and CMSCaller[18] have been utilized,they have not significantly impacted clinical practice.Our findings present an alternative protocol for patient classification,leveraging a 25-gene panel (TumorSec) and a three-gene RT-qPCR panel (TGF-β,β-catenin,and c-MYC).The selected genes play vital roles in the epithelial-mesenchymal transition,particularly TGF-β and β-catenin,which are specific to CMS4 (fibrotic)[19].Additionally,c-MYC was chosen due to its utility for identifying CMS2 (metabolic)[20].However,distinguishing between CMS2 and CMS3 remains challenging as they share genetic signatures and patterns of gene expression.
The relevance of classifying mCRC patients into CMS categories must be contextualized.Thus far,the selection of targeted therapies and the design of clinical studies have primarily relied on the identification ofKRAS,NRAS,andBRAFmutations and MMR expression analyses[7-8].However,incorporating knowledge of the CMS categories can offer significant advantages in both aspects.First,it can enhance the selection of targeted therapies,enabling a more personalized approach.Additionally,a better understanding of the CMS categories can lead to improved clinical study design,allowing for more tailored and effective treatments for patients with specific CMS profiles[6].For instance,CMS1,characterized by high lymphocytic infiltration and a worse prognosis,may benefit from aggressive therapeutic strategies such as combination triplet chemotherapy (FOLFOXIRI) and anti-angiogenic agents[21].Monodrug immunotherapy could also be beneficial for these patients given their high frequency of microsatellite instability-high tumours as demonstrated in the KEYNOTE177 study[22].Considering the high prevalence ofBRAFmutations,future studies should examine the efficacy of BRAF inhibitors for these patients[23].CMS2 and CMS3 share significant features and may respond to similar agents.For example,they may show sensitivity to anti-EGFR therapy,especially in CMS2 cases[24].However,CMS3 patients frequently developKRASmutations,primarily in exon 2,leading to constitutive activation of the mitogenassociated protein kinase pathway,associated with a poorer prognosis and response to standard treatment[25].CMS4,which carries the worst prognosis,calls for the development of new strategies targeting the epithelial-mesenchymal transition or the TGF-β pathway.CMS4 tumours also show better response to irinotecan-based treatments or antiangiogenic agents such as bevacizumab[26].
It is important to note that the classification of CMS can also predict the prognosis of patients with mCRC[4].While this study documented the overall survival of patients,there were no significant differences between groups,likely due to the low number of patients in each CMS category.Therefore,it cannot be established whether patients with different CMSs have different prognoses.
The principal innovation of this exploratory study lies in the establishment of a protocol for the classification of mCRC patients into CMS through RT-qPCR (TGF-β,β-catenin,and c-MYC) and a 25-gene NGS panel (TumorSec).Our results demonstrates that this combined approach has the potential to classify patients with mCRC into one of the four CMS categories in over 90% of cases.As there is currently no gold-standard for conducting this clinical-molecular classification,this approach may represent a significant advancement in the development of an optimal technique that could become the standard for these purposes.In the future,it is important to further explore CMS categories and incorporate this knowledge into clinical practice.While this protocol proposes a CMS classification scheme,prospective and large-scale studies are imperative to assessing whether this methodology truly influences therapeutic decisions for patients[5] and for validating the clinical utility of CMS categories[6].
CONCLUSION
In conclusion,we successfully classified mCRC patients into CMS categories using an RT-qPCR and NGS-based workflow.This approach opens avenues for tailoring therapies according to CMS subtypes,potentially leading to improved patient outcomes.
ARTICLE HIGHLIGHTS
Research background
Colorectal cancer is a heterogeneous disease;therefore,it is crucial to progress towards a molecular consensus classification in order to predict prognosis and therapy response.
Research motivation
The primary motivation is to progress towards a consensus molecular classification of metastatic colorectal cancer patients,to better guide targeted therapy.
Research objectives
The aim of this study is to classify a sample of metastatic colorectal cancer patients into consensus molecular subtypes using a reverse transcription -quantitative polymerase chain reaction polymerase chain reaction (RT-qPCR) and nextgeneration genomic sequencing (NGS) protocol.
Research methods
Patients with unresectable metastatic colorectal cancer who were undergoing systemic treatment with chemotherapy and/or targeted therapy.Molecular biology techniques were employed to analyse primary tumour samples from these patients.RT-qPCR was utilized to assess the expression of genes associated with fibrosis (TGF-β and β-catenin) and cell growth pathways.NGS using a 25-gene panel (TumorSec) was performed to identify specific genomic mutations.The patients were then classified into one of the four CMS categories according to the clinical consensus of a Tumour Board.
Research results
n=26 metastatic colorectal cancer patients analyzed.23% (n=6),19% (n=5),31% (n=8),and 19% (n=5) were classified as CMS1,CMS2,CMS3,and CMS4,respectively.Additionally,8% of patients (n=2) could not be classified into any of the four CMS categories.
Research conclusions
It is possible to classify patients with metastatic colorectal cancer into consensus molecular subtypes through RT-qPCR and NGS techniques.
Research perspectives
Prospective studies are needed to determine if this classification is useful and if it has an impact on predicting the survival of patients with metastatic colorectal cancer.
FOOTNOTES
Author contributions:González-Montero J led the study,wrote the manuscript,and created the figures and tables;González-Montero J,Burotto M,and Barajas O led the molecular classification of the patients;Valenzuela G,Toro J,and Marcelain K performed molecular biology procedures;Barajas O,Mateluna D,and Buen-Abad F recruited the patients.
Supported byAgencia Nacional de Investigación y Desarrollo de Chile,Fondo Nacional de Investigación en Salud (FONIS),No.SA20I0059.
Institutional review board statement:The study was reviewed and approved by the Medical Oncology Supervisor of Bradford Hill Clinical Research Center.
Informed consent statement:All study participants,or their legal guardian,provided informed written consent prior to study enrollment.
Conflict-of-interest statement:All the authors declare have not conflict of interest to declare.
Data sharing statement:No additional data are available.
STROBE statement:The authors have read the STROBE Statement—checklist of items,and the manuscript was prepared and revised according to the STROBE Statement—checklist of items.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Chile
ORCID number:Jaime González-Montero 0000-0003-0324-2948;Mauricio Burotto 0000-0002-7150-4033;Guillermo Valenzuela 0000-0002-8711-729X;Debora Mateluna 0009-0006-9030-7146;Florencia Buen-Abad 0009-0003-5914-6725;Jessica Toro 0000-0002-7813-1380;Olga Barajas 0000-0002-3013-2106;Katherine Marcelain 0000-0003-4018-6623.
S-Editor:Liu JH
L-Editor:A
P-Editor:Zhang XD
杂志排行

World Journal of Clinical Oncology的其它文章
- Comprehensive analysis of disulfidptosis related genes and prognosis of gastric cancer
- What should be the future direction of development in the field of prostate cancer with lung metastasis?
- Splenic lymphangioma masquerading as splenic abscess managed by laparoscopic splenectomy: A case report
- Hub genes and their key effects on prognosis of Burkitt lymphoma
- Treatment of patients with multiple brain metastases by isolated radiosurgery: Toxicity and survival