Ocular surface microbiota in patients with varying degrees of dry eye severity
2023-12-14XinRongZouPeiZhangYuanZhouYaoYin
Xin-Rong Zou, Pei Zhang, Yuan Zhou, Yao Yin
1Department of Ophthalmology, Fengcheng Hospital, Fengxian District, Shanghai 201411, China
2Fengcheng Branch, Shanghai Ninth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 201411, China
3Department of Ophthalmology, Huashan Hospital of Fudan University, Shanghai 200040, China
4Department of Ophthalmology, Gonghui Hospital, Jing’an District, Shanghai 200436, China
Abstract
● KEYWORDS: dry eye; microbiota; ocular surface; tear film break up time
INTRODUCTION
Dry eye (DE) is a globally prevalent disease[1]that damages the ocular surface (OS), seriously affecting vision and quality of life[2].Various components of the OS microenvironment maintain OS stability, whereas changes in components such as immune cells, matrix cells, hormones, and the microbiome, may lead to disruption of OS homeostasis[3].OS microbiota is another key component of the OS microenvironment, which maintains homeostasis and immune tolerance and eliminates pathogenic microorganisms[4].Alterations in OS microbiota can significantly affect the dynamic balance of OS microecology through quorumsensing[5].The ocular microbiota may be disrupted under certain conditions, such as DE, use of contact lenses,antibiotic treatment, and infections; additionally, impaired OS integrity in patients with DE promotes the pathogenic role of the microflora[6-9].Some pathogenic bacteria such asStaphylococcus aureusandPropionibacteriumare associated with DE[3,10].Hence, changes in OS microbial diversity in patients with DE deserve more research.
The new definition of DE includes its multifactorial nature with loss of tear film homeostasis central to its pathology[1].Recent epidemiological surveys have shown that shortened tear film break-up time (TFBUT) DE is the most common type of DE disease in clinical practice[11-13].Asia Dry Eye Society proposed DE diagnostic criteria according to DE symptoms and TFBUT<5s[14].Different severities of DE may lead to distinct clinical manifestations and consequences; thus,different remedial principles may be needed[1,15].Therefore, it is necessary to investigate DE according to its classification.The general objective of DE treatment is to restore OS microenvironment homeostasis.Restoration of normal OS commensal flora should be considered an indispensable part of DE therapy.Changes in the OS microbiota have been hypothesized to be involved in DE pathophysiology.However,previous studies did not classify DE according to disease severity[7,16-17].
In the present study, 16S ribosomal ribonucleic acid (rRNA)gene sequencing was performed to analyze the OS commensal bacteria profiles of patients with DE having different severities based on TFBUT values.We aimed to gain an improved understanding of the relationship between disease severity and microbial diversity.
SUBJECTS AND METHODS
Ethical ApprovalThe present study adhered to the ethical guidelines stated in the Declaration of Helsinki, and written informed consent was obtained from all participants.The study procedure was sanctioned by the Medical Ethics Committee of Fengcheng Hospital, Shanghai (approval number: FCYY-2019-TK322-1).
This single-center study included patients who sought treatment for DE-related symptoms at outpatient clinics of Fengcheng Hospital between December 2020 and June 2021.Prior to the investigation, two ophthalmologists collected participants’ medical history and enquired about subjective symptoms.Subsequently, participants underwent ocular examination using slit lamp microscopy; thereafter, other DE examinations were performed.
SubjectsThe study involved three groups of participants:1) control group (TFBUT≥5s), 2) mild DE (MDE) group(2s<TFBUT<5s), and 3) moderate-to-severe DE (MSDE)group (TFBUT≤2s).These groups had no significant differences in age and sex.The Asia Dry Eye Society 2016 guidelines were used to diagnose DE, which required the presence of subjective symptoms of DE and TFBUT<5s[14].Additionally, a cut-off value of TFBUT≤2s was applied in a large Norwegian cohort to investigate the severity of TFBUT and DE disease[18].
DE symptoms were defined as done in our previous studies, including dryness, foreign body sensation,redness, burning sensation, and sensation of heaviness in the eyelids.Participants who experienced one or more of the abovementioned symptoms often or persistently were categorized as positive for DE symptoms[19].
The exclusion criteria included the following ocular disorders which may affect tear production or quality[20-21]: 1) eyelid diseases: ectropion, entropion, ptosis, trichiasis, eyelid paralysis; 2) conjunctival disorders: palpebral fissures,pterygium, and conjunctivochalasis; 3) contact lenses use,history of ocular and periocular infection in the past three months, ocular or systemic antibiotic treatment within three months, ocular surgeries within six months, prior history of ocular chemical injuries, or use of ocular prescriptions or artificial tears; 4) systemic diseases and autoimmune diseases including diabetes, Parkinson’s disease, Grave’s disease,rheumatoid arthritis, Sjӧgren’s syndrome, and systemic lupus erythematosus.All enrolled patients were at least 18 years of age.
Tear Sample CollectionDE tests were performed in all subjects, including TFBUT measurement, Schirmer’s test without the use of anesthesia, and corneal fluorescence staining.The strips for Schirmer’s test (Jingming, Tianjin,China) were applied to the external third of the conjunctival sac in each lower eyelid and left for 5min.After removing the strips from both eyes, they were collected in sterilized centrifuge tubes as one specimen and immediately stored at−80°C for subsequent investigations as previously reported[21].When placing and collecting strips, sterile gloves were worn,and the hand that touched the strips had no contact with the lower eyelid skin.
Total Bacterial Deoxyribonucleic Acid ExtractionThe OMEGA Soil DNA Kit (M5635-02; Omega Bio-Tek,Norcross, GA, USA) was used to extract the entire bacterial DNA.The quality and quantity of the extracted DNA were evaluated using a NanoDrop NC2000 spectrophotometer(Thermo Fisher Scientific, Waltham, MA, USA).Agarose gel electrophoresis was also performed to confirm the integrity of the DNA.
16S rRNA Gene Amplicon SequencingThe bacterial 16S rRNAgenes V3-V4 region was amplified by polymerase chain reaction (PCR) using the forward primer 338F(5’-ACTCCTACGGGAGGCAGCA-3’) and the reverse primer 806R (5’-GGACTACHVGGGTWTCTAAT-3’).For multiplex sequencing, the primers used for PCR amplification of the bacterial 16S rRNA genes V3-V4 region were modified to include sample-specific 7 bp barcodes.Thereafter, Vazyme VAHTSTM DNA Clean Beads (Vazyme, Nanjing, China)were used to purify PCR amplicons.The Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA) was used to quantify the purified DNA.The PCR amplicons were individually quantified and combined in equal amounts before undergoing pair-end 2×250 bp sequencing on the Illumina NovaSeq platform with the NovaSeq 6000 SP Reagent Kit(500 cycles) at Shanghai Personal Biotechnology Co., Ltd.(Shanghai, China).
Sequence and Bioinformatics AnalysesMicrobiome bioinformatics analysis was conducted using Quantitative Insights Into Microbial Ecology software (QIIME 2).The raw sequence data were first demultiplexed using the demux plug-in in QIIME 2.The primers were then removed from the demultiplexed sequences using the cut-adapt plug-in.The quality-filtered sequences were processed using the DADA2 plug-in in QIIME 2, which includes denoising, the merging of paired-end reads, and the removal of chimeric sequences.The non-singleton amplicon sequence variants(ASVs) were aligned using MAFFT[22], and a phylogenetic tree was constructed using FASTTREE2[23].The ASVs were taxonomically classified using the classify-sklearn naïve Bayes taxonomy classifier in the feature-classifier plug-in, which was trained against the Greengenes_13 database.
The alpha-diversity and beta-diversity metrics were calculated using the diversity plug-in in QIIME2.The rarefaction threshold was set at 90 278 sequences per sample.QIIME2 software was used to determine alpha-diversity indices for each sample group separately, including the Chao1 index and Faith’s phylogenetic diversity (Faith’s PD) index for richness,Shannon and Simpson index for diversity, and three additional indices, involving Good’s coverage, Pielou’s evenness, and observed species.Box line plots were generated to compare the richness and evenness of ASVs among each sample group.The UniFrac distance metric was employed to analyze the beta diversity of microbial communities and investigate the differences in microbial community structures between groups.To examine the compositional profiles of species at the genus level, both principal coordinate analysis and nonmetric multidimensional scaling analysis were performed, which enable the visualization of similarities and differences in microbial community composition between different groups.Principal component analysis was used to identify patterns and relationships in the data along with outliers and anomalies at the genus level.To assess the significance of differences in microbial community structure between groups, analysis of variance using distance matrices and analysis of similarity were utilized.
QIIME2 was used to obtain composition and abundance tables at six taxonomic levels (phylum, class, order, family, genus,and species) for each sample.Bar graphs were generated to visualize the analysis results.Permutational multivariate analysis of variance was employed to assess the statistical significance of differences in the microbial community structure between groups.
QIIME2 and R packages (v3.2.0) were applied to analyze the sequence data.Linear discriminant analysis effect size(LEfSe) was exploited with default parameters to distinguish differentially abundant taxonomic categories among groups.Random forest analysis within QIIME 2 with default settings was utilized to develop predictive models by distinguishing between samples from different groups based on their microbial composition.Metabolic pathways and functions were predicted based on random forest ASV results using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2).
Statistical AnalysisClinical data analysis was conducted using SPSS (V.25.0, SPSS Science, Chicago, IL, USA).Descriptive statistics were provided for all outcome measurements, means and standard deviations were presented for continuous variables that met the normality assumption,and medians with Q1/Q3 were presented for continuous variables that did not meet this assumption.For categorical variables, frequencies and proportions were reported.Chisquare tests were operated to compare categorical outcomes,one-way analysis of variance (ANOVA) was conducted to compare continuous outcomes in compliance with a normal distribution (based on Shapiro-Wilk test), Kruskal-Wallis test was used to compare continuous outcomes with non-normal distribution, and the Nemenyi method was conducted for pairwise comparisons.The level of statistical significance was defined asP<0.05.
RESULTS
PopulationA total of 199 individuals were divided into three groups: the control, MDE, and MSDE groups with 61, 56, and 82 subjects, respectively.Table 1 displays the demographic and clinical characteristics of the participants.Our results revealed no significant differences in age and gender distribution among the three groups (P>0.05).The TFBUT and Schirmer’s test values were highest in the control group and lowest in the MSDE group, with a significant difference observed among the three groups.
Alpha DiversitiesAlpha diversity is a measure of the diversity and richness of species within a local or uniform habitat, which considers indicators such as species richness,diversity, coverage, and evenness.Significant differences in the following parameters were detected among the three groups:richness indices: Chao1 (P<0.001), Faith’s PD (P<0.001);diversity indices: Shannon index (P=0.0016), Simpson index(P=0.0086); Good’s coverage (P=0.0013); Pielou’s evenness(P=0.01).The control group displayed the highest richness(Chao1, Faith’s PD), and the MDE group showed the highest diversity (Shannon, Simpson), whereas the MSDE group had the lowest richness and diversity among the four indices(Figure 1A).
Beta DiversitiesBoth principal coordinate analysis(Figure 1B) and nonmetric multidimensional scaling analysis(Figure 1C) showed that the MSDE group had the lowest beta diversity.Principal component analysis at the genus level revealed that patients with DE in the MDE and MSDE groups presented different bacterial microbiome compositions from healthy participants in the control group (Figure 1D).There was a significant difference in beta diversity among these groups (ANOVA using distance matrices,P=0.001; Figure 1E),and analysis of similarity demonstrated the significant effect of DE on the diversity between groups (Table 2).

Figure 1 Alpha diversities and beta diversities of the three groups A: Alpha-diversity indices of the three groups.The P-values are the overall P-values of the Kruskal-Wallis test among the three groups; aP<0.05, bP<0.01, and cP<0.001.B–D: Beta diversity of the three groups.B:Principal coordinate analysis; C: Nonmetric multidimensional scaling analysis; D: Principal component analysis at the genus level; E: Difference exploration among the three groups via analysis of variance using distance matrices.PCo1: Principal coordinate 1; PCo2: Principal coordinate 2; NMDS1: Nonmetric multidimensional scale 1; NMDS2: Nonmetric multidimensional scale 2; PC1: Principal component 1; PC2: Principal component 2; MDE: Mild dry eye; MSDE: Moderate-to-severe dry eye.
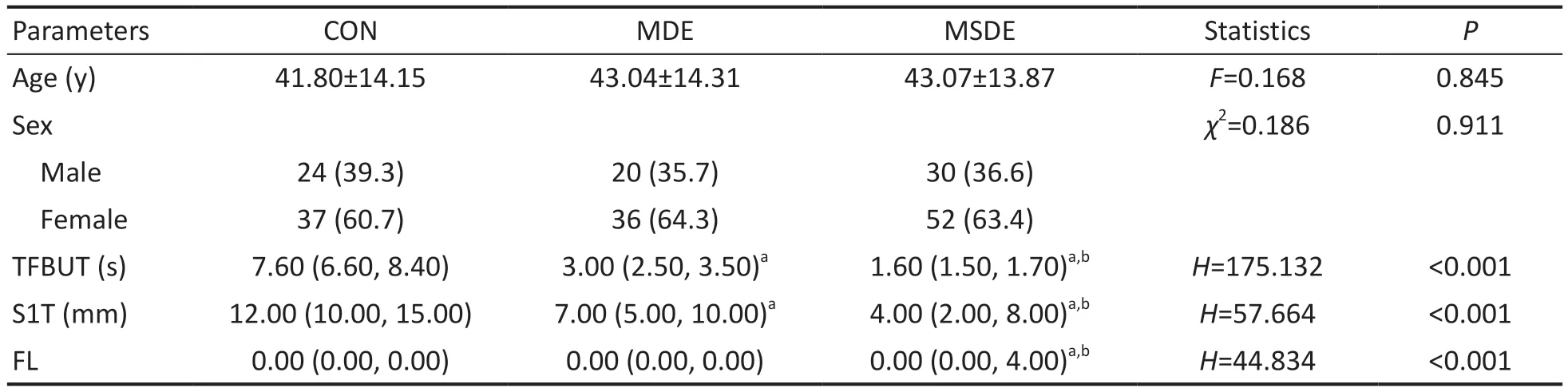
Table 1 Demographic and clinical characteristics: distribution of age, sex, TFBUT, Schirmer’s test results, and fluorescent staining
Bacterial Composition at the Phylum, Family, and Genus LevelsAt the phylum level, there was a gradual increase inProteobacteriafrom the control group to the MSDE group,whereasActinobacteria,Firmicutes, andBacteroidetesexhibited a decrease in abundance (Figure 2A).The composition of the top five phyla between the three groups showed significant differences (Figure 2D).
At the family level,Xanthomonadaceae,Oxalobacteraceae,Beijerinckiaceae,andAcetobacteraceaegradually increased from the control group to the MSDE group, whereasCorynebacteriaceae,Moraxellaceae,andStaphylococcaceaedecreased (Figure 2B).The composition of the top nine families in the three groups showed significant differences(Figure 2E).
At the genus level,CupriavidusandChelatococcussteadily increased from the control group to the MSDE group, whereasCorynebacterium,Lactobacillus, andStaphylococcaceae Staphylococcusdecreased (Figure 2C).EnhydrobacterandThermushad the highest relative abundance in the control and MSDE groups, respectively (Figure 2C).The composition of the genera with significant differences in the three groups is displayed in Figure 2F.
Linear Discriminant Analysis Effect SizeSpecies with significantly different abundances in different groups were presentedviaLEfSe.The taxa which had a greater influence on the difference between groups are shown in the LEfSe cladogram (Figure 3A).The representative top five taxa in the three groups at the family and genus levels were as follows:control group: family:Corynebacteriaceae,Lactobacillus,Moraxellaceae,Brucellaceae, andPropionibacteriaceae;genus:Corynebacterium,Lactobacillus,Enhydrobacter,Ochrobactrum,andPropionibacterium; MDE group: family:Pseudomonadaceae,Micrococcaceae,Sphingomonadales,Comamonadaceae,andNostocaceae; genus:Sphingomonas,Arthrobacter,Sporosarcina,Nesterenkonia,andDolichospermum; MSDE group: family:Xanthomonaceae,Oxalobacteraceae,Beijerinckiaceae,Thermaceae, andAcetobacteraceae, genus:Cupriavidus,Chelatococcus,Thermus,Chelativorans,andAllobaculum.
Identification of Species Differences and Marker Species using Random Forest AnalysisThe results from random forest analysis indicated significant differences (P<0.05) among the three groups (Figure 3B-3D).Notably,ProteobacteriaandThermiexhibited higher abundances in the MSDE group.Spirochaetes,Cyanobacteria,Acidobacteria, andChloroflexiwere highly abundant in the MDE group.At the family level,Pseudococcidae,Xanthomonadaceae,Beijerinckiaceae,Acetobacteraceae,Bacillaceae, andPlanococcaceaewere more abundant in the MSDE group.Pseudomonadaceaewas more abundant in the MDE group.At the genus level,Cupriavidus,Chelatococcus,Sphingopyxis,Tremblaya,Allobaculum,andArthrobacterwere more abundant in the MSDE group.

Table 2 Analysis of similarity for diversity between groups
The ASVs were found by random forest analysis (Figure 3E).The 10 most abundant ASV annotations in the MDE and MSDE groups are listed in Table 3.The ASVs were mainly classified into the following phyla and families:Proteobacteria:Pseudomonadaceae,Moraxellaceae, andPseudococcidae.
Predicted Metabolic Pathways and Functions based on Random Forest Amplicon Sequence Variants with PICRUSt2Prediction of metabolic pathways and functions was performed based on the top 20 random forest ASV results.Figure 4A shows the results of the differential analysis of KEGG metabolic pathways at Level 1.Differential analysis of Level 2 metabolic pathways belonging to human disease and metabolism in Level 1 pathways in the KEGG database are presented in Figures 4B and 4C.Human disease pathways included infectious diseases and neurodegenerative diseases with significant differences between the three groups.Metabolism pathways representing the metabolism of cofactors and vitamins, carbohydrate metabolism, lipid metabolism, and nucleotide metabolism exhibited significant differences and relative abundance between the three groups.
DISCUSSION
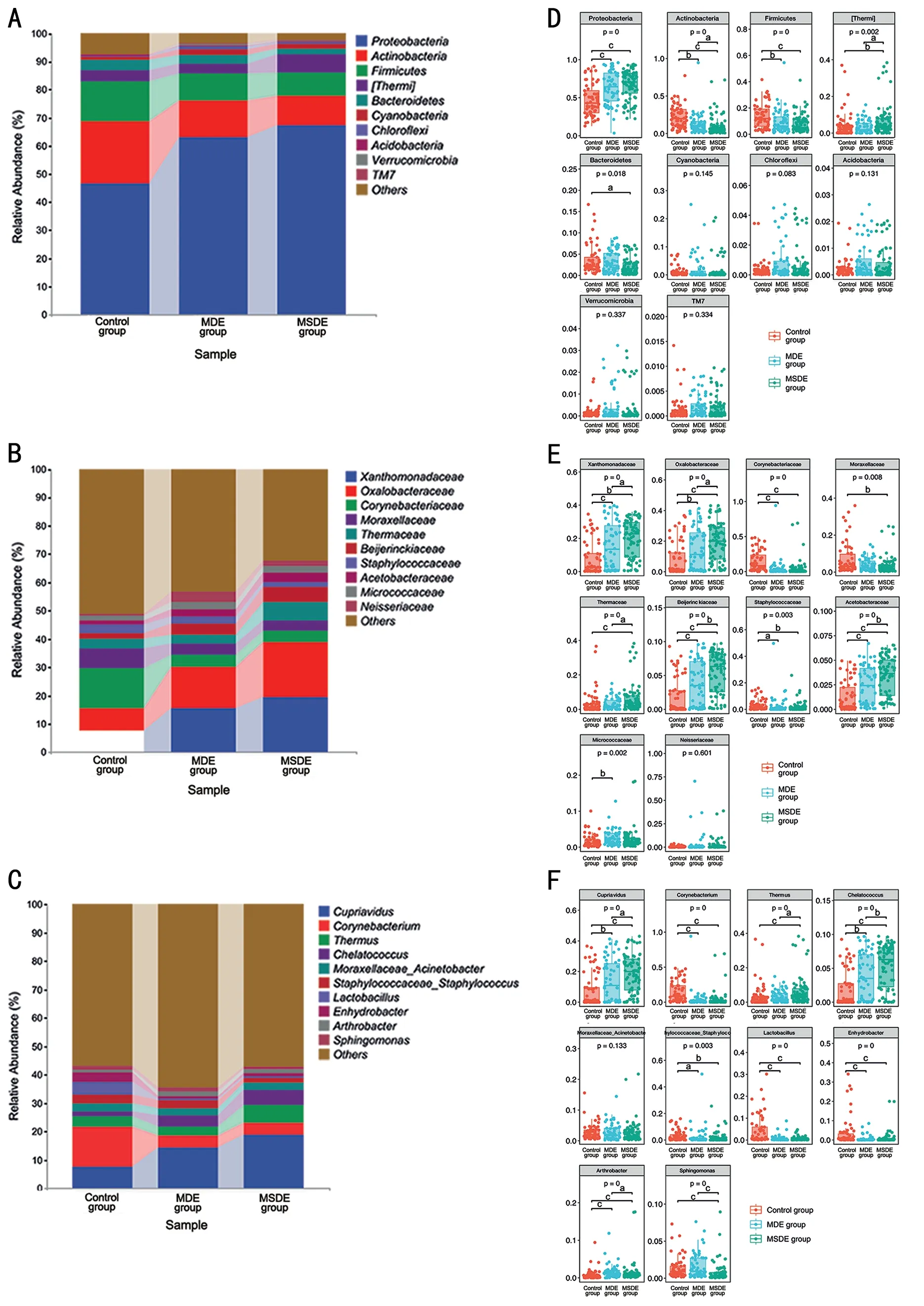
Figure 2 Bacterial composition of the three groups at the phylum, family, and genus levels A: Comparison of bacterial composition at the phylum level across the three groups.B: Comparison of bacterial composition at the family level in the three groups.C: Comparison of bacterial composition at the genus level in the three groups.D: Box plots for difference test at the phylum level in the three groups.E: Box plots for difference test at the family level in the three groups.F: Box plots for difference test at the genus level in the three groups.The P-values are the overall P-values of the Kruskal–Wallis test among three groups; aP<0.05, bP<0.01, and cP<0.001.MDE: Mild dry eye; MSDE: Moderate-to-severe dry eye.

Table 3 Classification annotation information about the ASVs of the marker group
Some studies have explored the commensal microbiota on the OS through various methods, including traditional microbial cultures and 16S rRNA gene sequencing[24-26].The microbiota plays a key role in maintaining the OS microenvironment balance under normal physiological conditions.The microbiota constitution can differ under certain conditions, such as DE, personal habits, rubbing eyes, antibiotic usage, use of contact lenses, infectious conditions, systemic diseases, and perioperative management[7,9,10,21,27-29].Thus, this study aims to investigate the OS microbiota composition of patients with DE having different severities based on TFBUT valuesvia16S rRNA sequencing using Schirmer’s strips.DE development was accompanied by corresponding changes in OS microbial components and abundance.These findings may indicate that DE development is accompanied by decreased microbial diversity and more homogeneous species composition.DE alters the frequency and density of specific bacterial groups.In this study, the MSDE group had the lowest richness and diversity in four indices of alpha diversity, while the control group had the highest richness as measured by the Chao1 and Faith’s PD indices.These findings share a similarity with Liet al’s[8]study, in that they both observed higher alphadiversity indices, specifically Shannon and Simpson, in the non-DE group compared to the DE group.In contrast, their study did not find any significant differences in the other two indices, including Chao1 and observed species, between the DE and non-DE groups[30].Additionally, the beta diversity of the microbial community decreased with increased DE severity(Figure 1B, 1C).Thus, the severity may be associated with a distinct assembly of the ocular bacterial community.Similarly,Anderssonet al[7]discovered reduced microbiota diversity among patients suffering from aqueous tear-deficient DE, and when compared to normal individuals, these patients exhibited differences in microbiota composition.Songet al[30]reported a significant difference in beta diversity among the three groups.However, in contrast to our findings, their control group exhibited a more centralized distribution of samples, whereas the samples in the two DE groups were more dispersed.The differentiation may be caused by the difference in the DE classification methods used in their study and the present study.A study examining oral microbial diversity in individuals with atrophic glossitis and healthy controls revealed that the former group exhibited lower levels of bacterial diversity than the latter[31].This study was analogous to our findings, which suggest a decrease in bacterial diversity in patients compared to healthy individuals.Therefore, it can be concluded that patients with more severe DE also exhibit lower levels of bacterial diversity.
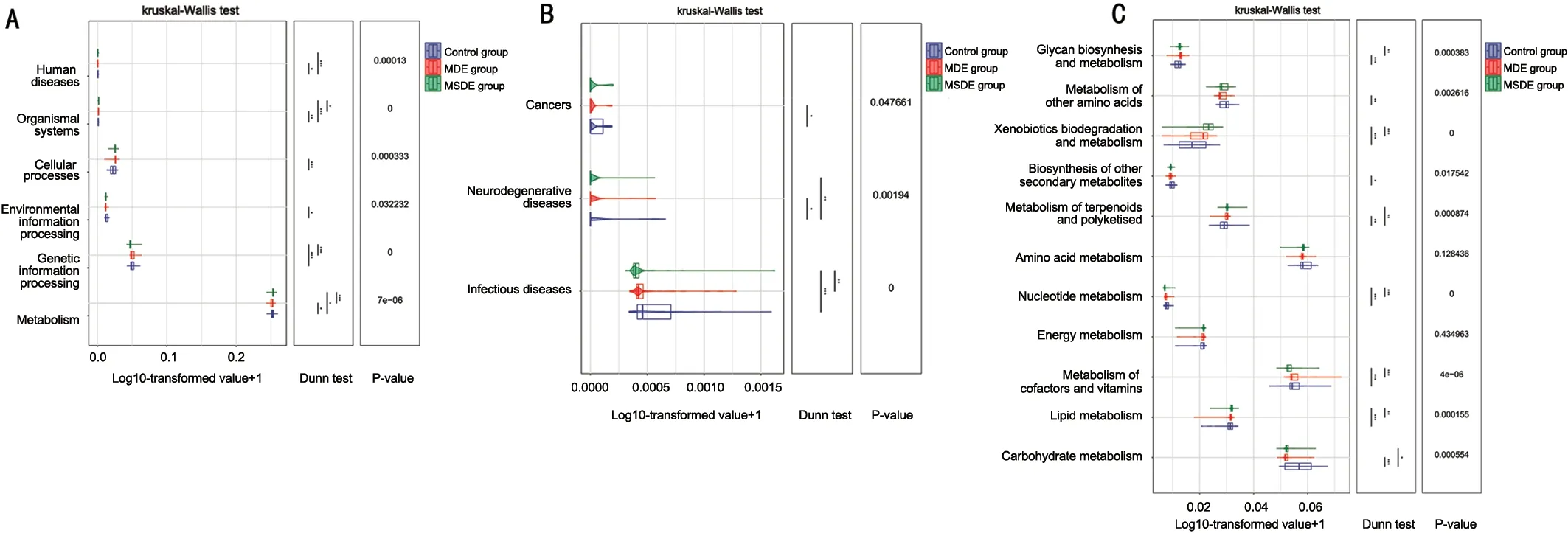
Figure 4 Prediction of metabolic pathways and functions using PICRUSt2 based on random forest ASV results A: Differential analysis of Level 1 metabolic pathways in the KEGG database.B: Differential analysis of Level 2 metabolic pathways belonging to human disease (Level 1)pathway in the KEGG database.C: Differential analysis of Level 2 metabolic pathways belonging to metabolism (Level 1) pathway in the KEGG database.PICRUSt: Phylogenetic investigation of communities by reconstruction of unobserved states; ASVs: Amplicon sequence variants; MDE:Mild dry eye; MSDE: Moderate-to-severe dry eye.
Alterations in bacterial composition were discovered among the three groups.At the phylum levels, Proteobacteria and Thermi abundance increased in the MSDE group compared with the MDE and control groups, whereasActinobacteriaandFirmicutesshowed the opposite trend in the same groups.In addition, the random forest analysis at the phylum level was consistent with the bacterial composition, that is,ProteobacteriaandThermishowed significant importance in the MSDE group.Proteobacteria,Actinobacteria,Firmicutes,andDeinococcus-Thermusare reported to be representative clones of human conjunctiva[26].Thermusis a widely distributed genus of thermophilic bacteria that can be isolated from both natural and man-made thermal environments[32].Based on the information presented, it is possible that the increased proportion of core taxa, such asProteobacteriaandThermi, in the MSDE group may negatively impact the microbiota homeostasis of the OS.Therefore, changes in the composition of these taxa in the OS could be closely related to DE development.
The discriminating genera in the control group were mainly harmless normal flora.For instance,Corynebacteriumis widely distributed in nature in the microbiota of animals and humans.Furthermore, they are the most common commensal flora that exists with their hosts due to their mostly harmless nature[33].In a study by Geet al[28],Corynebacteriumexhibited the highest relative abundance in the OS of healthy eyes from the control group.Moreover,Enhydrobacterwas also a core OS microbiota in most participants[7].In the MDE and MSDE groups, multiple pathogenic bacteria were detected.Pseudomonadaceaeshowed significant importance at the family level in the MDE groupviarandom forest analysis.The ASVs found in the current study were also mainly included inPseudomonadaceaeat the family level.Pseudomonadaceaeis a family of gram-negative bacteria, including the genusPseudomonas, which is pathogenic to humans[34].Pseudomonas aeruginosais a common pathogen that may induce severe ocular infection with possible vision loss[35-36], but thePseudomonadaceaedetected in this study with no classified information may belong to different species with dissimilar characteristics.Bacillaceae, which includes additional species with pathogenic potential,was vital at the family level in the MSDE group.Certain strains ofBacillus cereushave the potential to cause a range of infections, including localized wounds and ocular infections[37].Furthermore, a differential analysis of Level 2 metabolic pathways indicated that the infectious disease pathway exhibits significant differences.Thus, it can be inferred that the exacerbation of DE disease is accompanied by the enrichment of some pathogenic bacteria.Further investigations of the relationship between these pathogenic bacteria and DE are needed.
Some studies demonstrate that the effectiveness of supplementation with prebiotics or probiotics in reducing DE is associated with improved tear film function and restored OS microbiological activity in patients with DE[38-39].This may also be an option for future DE treatment, as there have been studies confirming the existence of a gut-eye axis[40-41].The OS microbiome is an extremely complicated issue that requires extensive research.Additionally, a more comprehensive longitudinal study may be considered for future analyses to examine how microbial composition changes after DE treatment and whether it returns to the control group level.
This study has some limitations.First, a separate group of subjects with TFBUT>10s was not set up; therefore,information about the OS microbiome of this group may have been missed.Second, as DE classification mainly depended on TFBUT values, the result does not fully reflect all types of DE conditions.Third, considering the geographical and sample size restrictions, there may be bias in the subjects that participated in the study.Different bacteria from other reports appeared in this study, and given that the OS is constantly exposed to bacteria from the external environment, it is highly probable that the microbiome present on the OS originates from external sources, such as water, earth, and air, as well as internal sources such as the body itself, including the lids,nasopharynx, obstetric canal, and skin[42].Therefore, different species with significantly different abundances found in each group in this study had their own regional characteristics, and further investigation of OS microbiota is needed.Lastly, aside from the identified bacteria, additional factors beyond the scope of the current study may have contributed to DE.For instance, latent infections such as infections withChlamydia trachomatisandUreaplasma urealyticumhave been observed in individuals with DE[43-44].Further exploration is necessary to comprehensively understand the causes of the complexity of DE.This study showed that patients with varying severities of DE had dissimilar bacterial diversities and OS microbial compositions.As DE severity worsens, microbial community diversity decreases, resulting in a more homogeneous ocular bacterial community structure that may be enriched with certain pathogenic bacteria.In this study, we investigated the microbial profile of patients with DE having different severities according to TFBUT values.Assessing alterations in the microecological OS environment across varying degrees of DE severity can provide valuable insights into the DE microbiome and potentially guide the development of effective treatment strategies.
ACKNOWLEDGEMENTS
The authors thank all the staffand participants of this study for their valuable skills and support.The authors thank Shanghai Personal Biotechnology Co., Ltd (Shanghai, China) for technical support.
Authors’ contributions:Zou XR and Zhang P are joint first authors.Zou XR designed this study.Zou XR and Zhang P analysed the data and drafted the manuscript.Zhou Y and Yin Y collected the data.All authors have read and approved the final manuscript.
Foundation:Supported byShanghai Municipal Health Commission (No.201940243).
Conflicts of Interest: Zou XR,None;Zhang P,None;Zhou Y,None;Yin Y,None.
杂志排行

International Journal of Ophthalmology的其它文章
- Dynamic tear meniscus parameters in complete blinking:insights into dry eye assessment
- Effects of diquafosol sodium in povidone iodine-induced dry eye model
- Morroniside ameliorates lipopolysaccharide-induced inflammatory damage in iris pigment epithelial cells through inhibition of TLR4/JAK2/STAT3 pathway
- Role of reactive oxygen species in epithelial-mesenchymal transition and apoptosis of human lens epithelial cells
- Electroacupuncture alleviates ciliary muscle cell apoptosis in lens-induced myopic guinea pigs through inhibiting the mitochondrial signaling pathway
- De novel heterozygous copy number deletion on 7q31.31-7q31.32 involving TSPAN12 gene with familial exudative vitreoretinopathy in a Chinese family