De novel heterozygous copy number deletion on 7q31.31-7q31.32 involving TSPAN12 gene with familial exudative vitreoretinopathy in a Chinese family
2023-12-14ShuangZhangHaiMingYongGangZouMeiJiaoMaXueRuiShangYingYangXunLunSheng
Shuang Zhang, Hai-Ming Yong, Gang Zou, Mei-Jiao Ma, Xue Rui, Shang-Ying Yang,Xun-Lun Sheng
People’s Hospital of Ningxia Hui Autonomous Region,Ningxia Medical University, Ningxia Eye Hospital, Ningxia Clinical Research Center on Diseases of Blindness in Eye,Yinchuan 750001, Ningxia Hui Autonomous Region, China
Abstract
● KEYWORDS: familial exudative vitreoretinopathy; copy number variation; copy number deletion; TSPAN12; longread sequencing
INTRODUCTION
Familial exudative vitreoretinopathy (FEVR; MIM:#133780) is a hereditary vitreoretinal disorder.It is characterized by abnormal incomplete vascularization of the peripheral retina.This can result in secondary neovascularization, which is prone to leakage or rupture.The condition can manifest in various clinical syndromes, ranging from mild peripheral avascularity with normal visual acuity to severe retinal detachment, ultimately leading to blindness.FEVR can associate with intellectual disabilities[1-2], and is reported to be autosomal dominant (AD), autosomal recessive inheritance (AR), or X-linked inheritance trait, and genetically associated with the genes ofFZD4[3],LRP5[4],TSPAN12[5],NDP[6],KIF11[7],ZNF408[8],RCBTB1[9],CTNNB1[10], andJAG1[11].While point gene variations in nucleic acids have commonly been implicated in the disease, recent studies have also identified copy number variants (CNVs) as contributing factors.CNVs can alter gene dosage, modify the 3D architecture of the genome, or even result in the formation of chimeric genes[12].
CNVs, which are DNA segments larger than one kilobase (kb),could present variable copy numbers compared to the reference genome.They are an important aspect of genomic diversity,similar to single nucleotide polymorphisms (SNP), and may not be associated with any disease phenotypes[13].However, in the cases of FEVR, six genes (FZD4[14],LRP5[12],TSPAN12[12],NDP[15],KIF11[12],andCTNNB1[14]) have been reported to be affected by CNVs.Understanding the pathology of CNVs is crucial in determining the genes responsible for the disease and identifying dosage-sensitive genes for further investigation.However, many of these reports lack precise breakpoint information, making it difficult to analyze the relationship between phenotypes and the specific genes affected by the CNV region.For instance, individuals with heterozygous deletion of the entireTSPAN12gene can exhibit clinical features such as cleft lip, dysmorphic ears, and death at an early age, or only milder FEVR phenotype[16].It is unfortunate to miss out on analyzing other critical genes within the CNVs region that could be linked to these syndromes.Detecting precise breakpoints using the commonly used whole-exome sequencing (WES) technique is challenging for large segments with copy number deletion or duplication, compromising the reliability of the results.Therefore, the discussion needs to focus on finding appropriate methods to resolve CNVs and determining the critical genes within the entire CNV region that are associated with specific syndromes.
Here we reported the identification of a heterozygous 4.5 Mb copy number deletion on chromosome 7q31.31-7q31.32, which includes the entireTSPAN12gene, in a family from Northwest China exhibiting the FEVR phenotype.In comparison to previous reports of heterozygous wholeTSPAN12deletions,we discovered ade novopathological copy number deletion located on 7q31.31-7q31.32 and confirmed the breakpoint as del(7)(q31.31q31.32)chr7:g.119451239_123956818del.Accurate molecular diagnosis of genetic disorders plays a crucial role in understanding the pathogenesis of complex syndromes.This is particularly important for families affected by hereditary diseases, as it enables the identification of pathogenic variants and provides valuable insights into the correlation between phenotype and genotype.
SUBJECTS AND METHODS
Ethical ApprovalOur study adhered to the Declaration of Helsinki and followed the collection guidelines for human genetic disease specimens issued by the Ministry of Health of China.It was approved and reviewed by the Ethics Committee on Human Research at People Hospital of Ningxia Hui Autonomous Region.The approved number is 2021-NZR-136.Written informed consent was received from each participant or his/her legal guardians prior to their participation.
Clinical Examinations, Diagnosis, and Grading of Familial Exudative VitreoretinopathyThree patients (individuals of Figure 1A, I:1, II:1, and II:2) with FEVR, as well as one unaffected family members (individuals of Figure 1A, I:2),from a Chinese family were recruited for both genetic and clinical tests.The parents of the patients denied exposure to toxicants and any adverse personal history.All family members underwent comprehensive ophthalmic examinations including best-corrected visual acuity (BCVA), refractometry(Topcon KR8100, Topcon Inc., Japan), slit-lamp examination,color fundus photography (Heidelberg Engineering GmbH,Heidelberg, Germany), optical coherence tomography (OCT)examinations (HD-OCT4000, Carl Zeiss Meditec, USA) and fundus fluorescein angiography (FFA, Heidelberg Engineering GmbH, Heidelberg, Germany).Visual acuity values were recorded using logMAR data.Prior to the fundus examination,bilateral pupil dilation was achieved using 0.5% compound tropicamide eye drops (Santen Pharmaceutical Co., LTD.M605191, Japan).The proband also underwent systemic clinical evaluations, including Denver Developmental Screening Test.
The clinical diagnosis of FEVR disease was based on the following three criteria: the presence of at least one eye with peripheral retinal avascular area; no history of premature delivery or oxygen inhalation, excluding neonatal retinopathy(ROP) and Norrie disease; the presence of any degree of peripheral retinal avascular area, increased branching, brushlike border, vitreous retinal or macula traction, a peripheral fibrovascular mass or fibrous proliferation, subretinal exudation or retinal neovascularization.
The severity of FEVR was graded according to the following criteria: stage 1: avascular area of the peripheral retina; stage 2:avascular area of the peripheral retina with neovascularization,without/2A or with/2B the appearance of exudate or angiographic appearance of leakage in the late phase; stage 3:subtotal retinal detachment that does not involve the macular area, without/3A or with/3B the appearance of exudate; stage 4: subtotal macula-involving retinal detachment, without/4A or with/4B the presence of exudation; stage 5: total retinal detachment divided into an open funnel type and closed funnel type.
Whole Exome AnalysisWES was conducted on the four individuals (Figure 1A, I:1, I:2, II:1, and II:2) from this family in order to identify the mutation responsible for their disease.Genomic DNA samples from each patient were sheared into 300-500 bp fragments and then ligated with Illumina Y-shaped adaptors.After purification using Agencourt AMPure SPRI beads, the samples were amplified using ligationmediated polymerase chain reaction (PCR).The SeqCap EZ Hybridization and Wash kit (Roche NimbleGen, Madison,WI) covering 44.1 megabases (Mb) was then employed for the enrichment of over 20 000 genes per the manufacturer’s protocols.

Figure 1 Validation the copy number deletion A: Pedigree information.In this pedigree, filled symbols indicate affected patients with FEVR,the unfilled symbols indicate unaffected individuals.Squares represent males, and circles represent females.The letter “M” represents copy number deletion, and “+” indicates the normal allele.The arrow points to the proband (II:1) who exhibits copy number deletion of del(7)(q31.31q31.32)chr7:g.119451239-123956818del.B: Long-read technology (ONT) shows aberrant decrease reads at 7q31.31-31.32.C: The copy number deletion encompasses 21 genes with TSPAN12 included, spans approximately 4.5 Mb.D: Sanger sequencing identified the breakpoint of the copy number deletion, which spans from position g.119451239 to g.123956818.E: PCR products including the recombination site of the breakpoint, it revealed a single band at 1000 bp in the three patients (C, I:1, I:2, II:1), and none in the mother (C, I:2 ).FEVR: Familial exudative vitreoretinopathy; ONT: Oxford Nanopore sequencing technology; PCR: Polymerase chain reaction.
The post-capture libraries were quantified using Pico green assay and sequenced on an Illumina Hiseq 2000 machine, as indicated previously[17].Variants with minor allele frequencies(MAFs) greater than 0.5% were filtered based on healthy population frequency databases including dbSNP, 1000 Genome Project (1000G, http://www.internationalgenome.org/data), the Exome Aggregation Consortium Browser (ExAC,http://exac.broadinstitute.org), the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org/).Novel and rare variants with MAF less than 0.5% were classified and analyzed for pathogenicity according to the guidelines set by the American College of Medical Genetics and Genomics(ACMG)[18].
Oxford Nanopore Sequencing TechnologyFor Oxford Nanopore Sequencing Technology (ONT), genomic DNA isolation was performed using the same method as described for WES.The DNA was then purified with a 1×reaction using Agencourt Ampure XP Beads (NC9959336, Fisher Scientific,Hampton, NH, USA).The DNA was then treated with the NEB Next Ultra II End-Repair/dA-tailing Module (NEB E7546S, New England Biolabs, Ipswich, Massachusetts, USA)to repair any damaged template DNA.Library preparation was carried out according to the manufacturer’s protocol, and sequencing was performed for 48h using an MK1B MinION.The sequencing and base calling processes were conducted using MinKNOW version 1.1.21 and Metrichor version 1.125(ONT, Oxford, UK).The native Fast5 files were converted to FASTQ files using Poretools[19].
Quantitative Real-Time Polymerase Chain Reaction Verification ofTSPAN12HaploinsufficiencyQuantitative real-time polymerase chain reaction (QPCR) amplified the region of interest on the target gene and the control was performed on the samples that meet the quality standards.The fluorescence was used to monitor the PCR process in real-time, allowing for relative quantitative analysis of each sample DNA.The exon 8 of theTSPAN12gene was amplified using theTTLL5andSPATA7genes as endogenous references.Primers were designed specifically for the exon 8 (Fw: 5’-TGCTCTGGGCTCTGTATTATGA-3’, Rv: 5’-CAGGTGCTGAGAGTTGTCATTC-3’).The beginning step in the PCR is performed at 95℃ for 10min, then came to 40cycles of denaturation stage at 95℃ for 10s, annealing at 60℃for 1min.The next stage is the melt curve stage, performed at 95℃ for 10s, 65℃ for 1min, 95℃ for 15s, and last, for the cooling stage, performed at 40℃ for 10s.
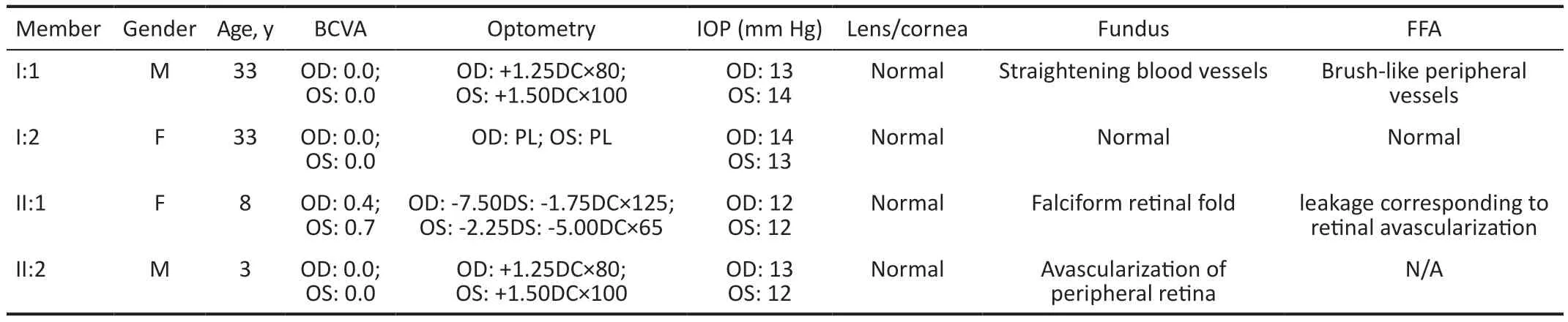
Table 1 Clinical details of the family members
Polymerase Chain Reaction and Sanger SequencingPCR and Sanger sequencing were also performed to validate the breakpoint regions of CNVs.Primers were designed as Fw: 5’-GCTGCTCCATACCTGTATCAC-3’, Rv:5’-CATAAGTTCCAAGAGTACATAGAC-3’, including the recombination point.PCR was carried out using a LongAmp Taq PCR Kit (E5200S; NEB, Ipswish, MA, USA).Totally 50-100 ng gDNA was used as PCR template (conditions: 96℃for 5min followed by 35 cycles of 96℃ for 30s, 60℃ for 30s,and 72℃ for 4min, with a final extension at 72℃ for 4min).PCR products purification, library construction, sequencing,and data analysis were the same as described in WES.Sanger sequencing was used to validate the breakpoint junctions.
RESULTS
Clinical FeaturesThe clinical features of FEVR were observed in three patients (Figure 1A, I:1, II:1, and II:2) from a family with a copy number deletion on the 7q31.31-7q31.32 region.The details of the patients’ clinical data are summarized in Table 1.Based on a comprehensive clinical examination, an AD inheritance pattern was identified in this family.(Figure 1A).The proband (Figure 1A, II:1), who was referred to our hospital at the age of 8 presented with decreased visual acuity in both eyes and esotropia.She had normal mental development and did not exhibit any syndromes of autism.The Denver Developmental Screening Test results were normal.The BCVA was measured as 0.4 in the right eye and 0.7 in the left eye.Both the corneas and lens appeared normal.Retinal photography revealed a falciform fold extending from the optic disc to the temporal retina.(Figure 2A, 2B, white arrows),accompanied by macula dragging in both eyes, avascular areas with fiber proliferation (Figure 2A, yellow arrows).FFA demonstrated temporal peripheral retinal nonperfusion zones and the presence of abnormal new blood vessels (Figure 2E, 2J, red arrows), as well as fluorescence leakage (Figure 2D, 2E, yellow arrows).Peripheral fibrovascular mass with hyperfluorescence indicated an abnormal increase in retinal vascular branches (Figure 2I, yellow arrows).The upper and lower vascular arches showed sharpened angles, pulling towards the temporal side (Figure 2C, red arrows).Optic disc leakage was observed in both eyes (Figure 2G, yellow arrows).OCT scans demonstrated a flatter central macula in both eyes (Figure 2K shows the right eye, 2L shows the left eye).According to Trese’s Staging System, the proband exhibited stage 2A symptoms without exudate.
The asymptomatic father (Figure 1A, I:1) exhibited a normal anterior segment.His fundus photography revealed a regular posterior pole of the retina (Figure 3A, 3B), but the avascularized area of the peripheral retina in both eyes (Figure 3A, 3B, white arrows).FFA demonstrated brush-like peripheral vessels with fluorescence leakage (Figure 3F, 3I, 3J, red arrows) and retinal neovascularization without exudate (Figure 3C, 3H, yellow arrows), which were considered as typical signs of FEVR stage 2A, even though he had normal visual acuity in both eyes.FFA image also demonstrated anomalous vascularization, supernumerary vascular branching in areas of vascular-avascular junctions (Figure 3D, 3E, brown arrows),and the no perfusion areas (Figure 3G, 3J, green arrows).The OCT shows no apparent abnormalities in morphology and thickness of the macular (Figure 3K, 3L).
The younger brother of the proband (Figure 1A, II:2), whose parents refused to undergo FFA, presented with normal visual acuity and anterior segments, but had abnormal avascular peripheral retinal vessels.The proband’s mother (Figure 1, I:1)showed normal visual acuity, the anterior segment and fundus examination.However, the proband had an earlier onset of the disease, and her visual acuity decreased significantly and rapidly progressed.
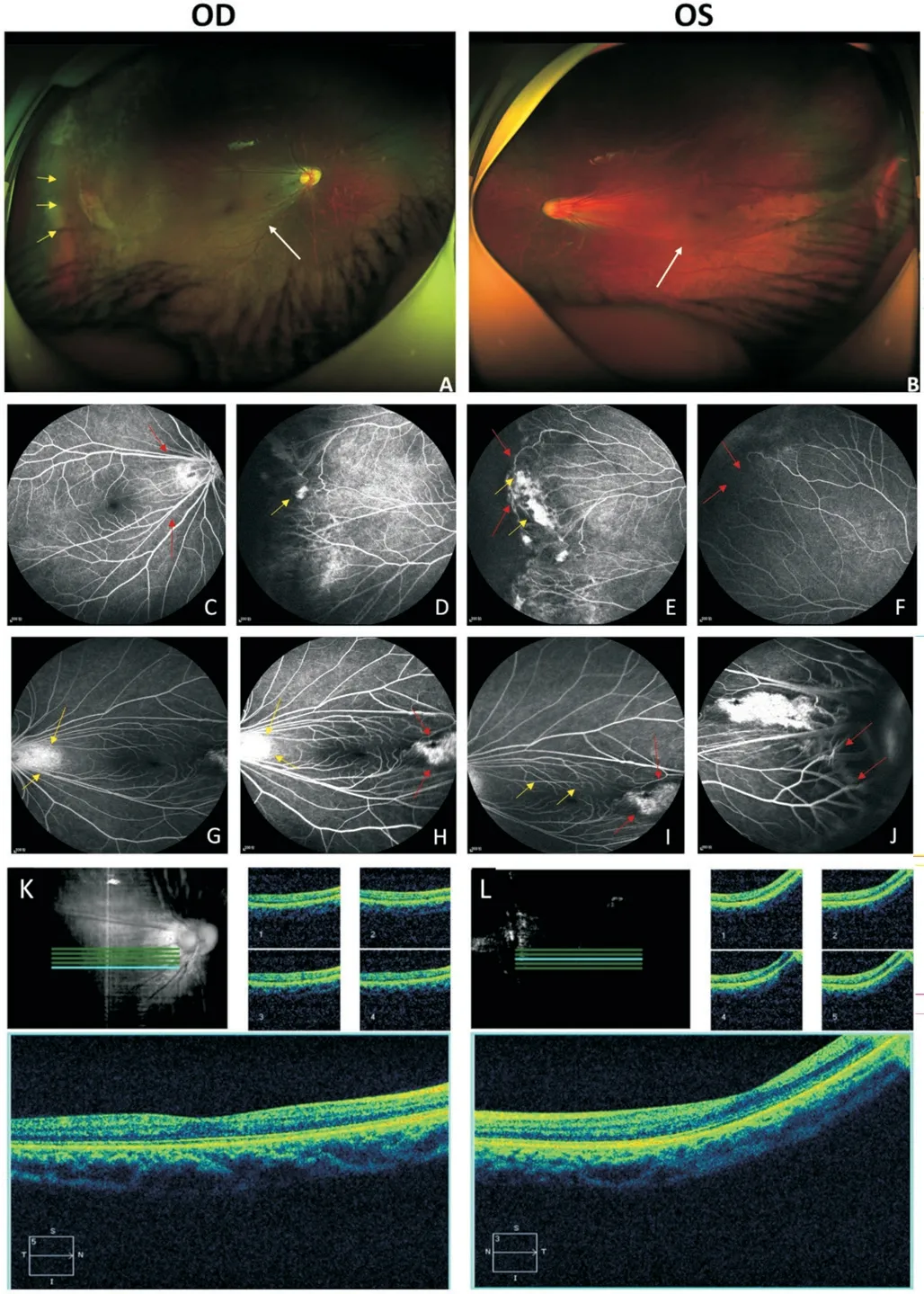
Figure 2 Clinical observations of the proband (II:1) Fundus photography exhibited falciform retinal fold and macula dragging in both eyes (A, B white arrows), as well as an avascular area with fiber proliferation (A, yellow arrows).FFA exhibited temporal peripheral retinal nonperfusion zones (F, red arrows), abnormal new blood vessels leakage (E, J, red arrows), fluorescence leakage (D, E, yellow arrows), peripheral fibrovascular mass with hyperfluorescence (H, I,red arrows), and an abnormal increase of retinal vascular branches(I, yellow arrows), pulling towards the temporal side, sharpened angle of upper and lower vascular arches (C, red arrows), and optic disc leakage (G, H, yellow arrows).OCT scan showed a flatter central macula in both eyes (K shows the right eye and L shows the left eye).FFA: Fundus fluorescein angiography; OCT: Optical coherence tomography.
Genetic FindingsTo identify the genetic cause of FEVR in this family, we performed WES on all the family members(Figure 1A, I:1, I:2, II:1, and II:2).WES analysis detected a potential deletion on chromosome 7 in the proband (Figure 1A, II:1), the affected father (Figure 1A, I:1), and the younger brother (Figure 1A, II:2).Subsequently, CNV analysis on the WES data using Exome Depth revealed that the proband(Figure 1A, II:1) and the affected father (Figure 1A, I:1) had the same large deletion on chromosome 7 in minus one copy ratio, which was approximately 4 Mb in size (Figure 4).To identify the exact breakpoint of the CNV deletion in the FEVR patients, we performed further genotypical and phenotypical analyses.The resulting call set of the proband from ONT revealed 212 copy number deletions, 105 copy number loss(the deletion is more obvious than the loss), 78 copy number gains, and 3 copy number amplification (the amplification is more evident than the gain).By comparing these results with the WES and CNV-seq findings, we were able to pinpoint the breakpoint site.This analysis confirmed a heterozygous copy number deletion affecting 21 genes (Figure 1B, 1C).The left breakpoint was located at (hg38) Chr7:119451239,while the right breakpoint was at chr7:123956818.The copy number deletion, chr7:g.119451239-123956818 covered a region of 4 505 580 bp that was not previously reported in gnomAD.The 21 genes involved in the heterozygous copy number deletion wereKCND2,WASL,CADPS2,HYAL4,IQUB,ASB15,PTPRZ1,FAM3C,CPED1,SPAM1,NDUFA5,SLC13A1,FEZF1,AASS,WNT16,ING3,LMOD2,TAS2R16,RNF148,RNF133,TSPAN12.Table 2 lists 15 genes, along with their OMIM numbers and functions.
Figure 3 Clinical observations of the father (I:1) Fundus photography exhibited an avascularized area (white arrows) in the peripheral retinal (A, B).FFA demonstrated brush like peripheral vessels with fluorescence leakage (F, I, J, red arrows), retinal neovascularization(C, H, yellow arrows), no perfusion area (G, J, green arrows) and supernumerary vascular branching in areas of vascular-avascular junctions (D, E, brown arrows).OCT is normal in both eyes (K shows the right eye, L shows the lefteye).FFA: Fundus fluorescein angiography; OCT: Optical coherence tomography.

Figure 4 The results of CNV analysis conducted on FEVR patients The analysis conducted was performed using Exome Depth and revealed that both the proband (A) and her father (B) had the same large deletion on chromosome 7, resulting in a minus 1 copy ratio.This deletion spans approximately 4 Mb (short orange line) and is represented by short orange line.C: The TSPAN12 CNV-seq analysis of four family members affected by FEVR.The x-axis indicates the exon 8 of TSPAN12, as well as two control gene regions (SPATA7 exon 6 and TTLL5 exon 14).The y-axis represents the number of copies detected in each specific region.Asterisks highlight the heterozygous deletion of the target gene, TSPAN12, in comparison to the reference genes compared TTLL5 and SPATA7. CNV: Copy number variation; FEVR: Familial exudative vitreoretinopathy.

Table 2 The genes detected in the 4.5 Mb CNV deletion located in 31.31-7q31.32
The heterozygous deletion ofTSPAN12gene in the affected individuals was further confirmed by QPCR using primers specific toTSPAN12exon 8.The proband (Figure 1A, II:1),the affected father (Figure 1A, I:1), and her younger brother(Figure 1A, II:2) showed half of the copy number compared to the healthy mother forTSPAN12exon 8.Conversely, the control genomic regions,TTLL5exon 14 andSPATA7exon 6, showed equal copy numbers in all individuals (Figure 4C).Therefore, the heterozygous deletion involving theTSPAN12gene was considered the pathological cause of the patients with varying disease phenotypes.Additionally, segregation analysis confirmed the presence of the unaffected mother on the allele,confirming that the deletion originated from the father.
To further delineate the deletion, targeted PCR amplification and Sanger sequencing were performed, leading to the identification of the same junction product.However, refining the deletion through iterative PCR was not possible due to the large size of the deletion, which exceeded the amplification range of rudimentary PCR.Primers were designed for the entire family, incorporating the recombination site of the breakpoint.The three patients showed a single band of approximately 1000bp (Figure 1E, I:1, I:2, II:1), while the mother presented none (Figure 1E, I:2).
DISCUSSION
We herewith report on a Chinese family affected by FEVR with a novel 4.5 Mb copy number deletion at 7q31.31-31.32.Two siblings (Figure 1A, II:1, II:2) and their father (Figure 1A) from Northwest China were found to have this deletion.Previous studies have indicated that deletions in the 7q31.31-31.32 region are associated with retinal angiogenesis and mental development[20].The existing literature correlatedcopy number deletions on 7q31.31-31.32 with a range of developmental phenotypes.We compiled a list of phenotypes associated with copy number deletions in this region, ranging from global developmental delay to specific developmental or morphological phenotypes, using the UCSC database.(UCSC,http://genome.ucsc.edu; Table 3).The pathogenic effect of 7q31.31-31.32 copy number deletion involved the phenotypes of speech and language disorders due to the deletion of theFOXP2gene, or intellectual disability and developmental delay due to the deletion of theIMMP2Lgene[20-21].In addition,a previous report described a patient with a 5.4-Mb deletion in the 7q31.31 region involving theCADPS2andTSPAN12gene, who exhibited bilateral persistent hyperplastic primary vitreous (PHPV) apart from autism spectrum disorders (ASD)phenotype[22].
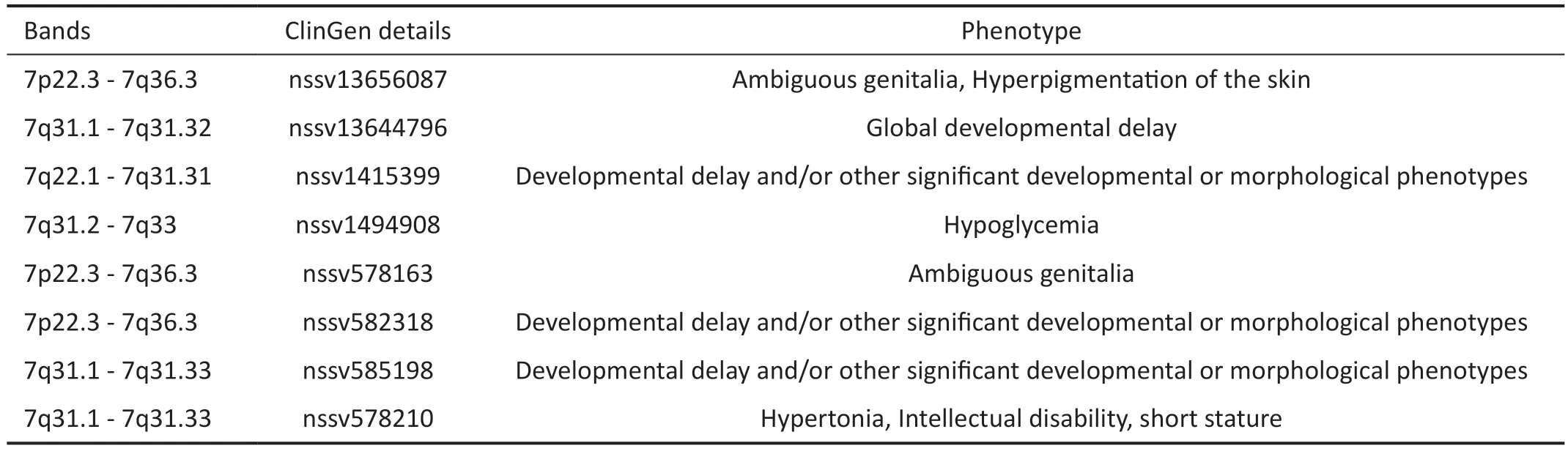
Table 3 The details of copy number deletion involving 31.31-7q31.32 listed on UCSC
For our study the variant region is located at 7q31.31-31.32,del(7)(q31.31q31.32)chr7:g.119451239_123956818del, and comprises 21 protein-coding genes.These genes includeTSPAN12,KCND2,WASL,CADPS2,HYAL4,IQUB,ASB15,PTPRZ1,FAM3C,CPED1,SPAM1,NDUFA5,SLC13A1,FEZF1,AASS,WNT16,ING3,LMOD2,TAS2R16,RNF148,RNF133.Table 2 lists the 15 genes with OMIM number and function.Our patients only present a mild FEVR phenotype attributed to theTSPAN12heterozygous copy number deletion.It’s worth noting thatKCND2andCADPS2,which have been reported to correlate with autism features, did not appear in our patients.Additionally, the deletion contains 3 OMIM disease genes, they areFEZF1gene associated with autosomal recessive hypogonadotropic hypogonadism 22, with or without anosmia (MIM: #616030),AASSgene with autosomal recessive hyperlysinemia, (MIM: #238700) andTAS2R16gene with AD beta-glycopyranoside tasting (MIM: #617956).However, these genes were not subjected to a special analysis as a second correlated gene because the affected individuals did not exhibit any clinical signs of the associated disorder.Combining the phenotype and genotype, haploinsufficiency ofTSPAN12was considered the main pathological cause due to its role in FEVR.
TheTSPAN12gene encodes a 305 amino acid quadruple transmembrane protein, which belongs to the Tetraspanin family.TSPAN12 contains four transmembrane domains connected by three loops, including a small extracellular loop,a large extracellular loop, and a tiny inner loop.The large extracellular loop includes a conserved Cys-Cys-Gly sequence(the CCG motif) and two additional cysteines, which form disulfide bonds and are crucial for protein folding.TSPAN12 is involved in a series of membrane-related activities, including cell adhesion, cell proliferation, and signal transduction[5].In the retina, TSPAN12 is selectively expressed in the retinal vasculature and acts as a receptor for Norrin.It binds with FZD4 and LRP5 to form a protein complex, playing a central role in retinal vascularization by regulating Norrin (NDP)-induced β-catenin signaling transduction and regulation but not Wnt- induced β-catenin signaling.The protein interacts with Norrin or LRP5 to enhance the polymerization reactions of the Norrin/FZD4/LRP5 complex in the retina.Lack of TSPAN12 may lead to reduced Norrin/FZD4/LRP5 signaling,which controls angiogenesis.This signaling reduction can be rescued by TSPAN12 overexpression, even though direct binding with Norrin and FZD4 was not detected[5,23].Therefore,a TSPAN12-related product has been designed as a treatment for vasoproliferative retinopathy.Furthermore, a TSPAN12 antibody serves as a delivery vehicle for endothelial-specific therapeutics and shows strong support for physiologic revascularization in hypoxic retinal tissue compared to anti-VEGF treatment.TSPAN12 activity may also present a promising target for encouraging physiologic revascularization in avascular areas in conditions like diabetic retinopathy or vein occlusion, yet an unsolved problem[24].
The precise breakpoint of the CNVs provides crucial information for narrowing down the susceptible genes associated with the observed phenotypes.However, it is important to consider that phenotypes can also be influenced by the genetic background in heterozygous individuals and microenvironmental factors in the eyes.Therefore, when analyzing phenotypes, it is essential not to neglect the role of each gene contained in the CNVs.Mutations in theTSPAN12gene have been found to count for 5.6% to 8.0% of FEVR patients and are common (12.8%)in patients with asymptomatic mild FEVR[25].A total of 12 FEVR patients with CNVs involving theTSPAN12gene have been reported so far (Table 4)[16,25-27], with 4 out of 12 cases showing a complete deletion of the entireTSPAN12gene.Among these cases, there was only one affected individual who exhibited additional symptoms such as cleft lip, lowset dysmorphic ears, and died at the age of 20mo, along with FEVR.The remaining three cases showed only mild phenotypes.Without knowing the precise breakpoint, it is difficult to determine the contribution of theTSPAN12gene to the syndrome.The large CNV region often spans several mega bases and encompasses multiple genes, making it challenging to identify the specific gene that should be prioritized as a target for further investigation into the underlying pathological mechanisms.However, only in one family, four members with a copy number deletion of theTSPAN12gene involving exon 7 were included, and the precise breakpoint was determined using Whole Genome Sequencing (WGS).Interestingly, the cases in our study with the entireTSPAN12gene deletion included showed a mild phenotype, providing support for the hypothesis that the dosage effect of theTSPAN12gene contributes to the development of mild syndrome.This is in contrast to the CNV-associated phenotype observed in theNDPgene,which leads to congenital blindness with total retinal detachment[15].Furthermore, CNVs can include entire functional genes within coding and regulatory regions, thereby altering the expression levels of target genes through genedosage effects and ultimately increasing or decreasing the disease risk.Additionally, genes located in noncoding regions of the chromosome can have enhancer or suppressor roles through long-range regulatory effects, indirectly affecting the expression levels of downstream targets.Thus, further studies investigating the breakpoint of the CNVs associated with FEVR are warranted to gain a better understanding of their function.Genome sequencing is an effective tool for evaluating the significance of novel CNVs and determining their exact breakpoints.Routine genetic workups like WES are unable to reliably detect large CNVs since they are often located in noncoding regions and exceed the short reading span of WES.Therefore, it is crucial to adopt an appropriate approach to detect CNVs in FEVR.In our study, we initially used high throughput and efficacy WES to locate CNV on chromosome 7q31.However, WES failed to detect the precise site of the breakpoint.To overcome this limitation, we employed longread genome sequencing ONT, which provided accurate breakpoint detection.The accuracy of the breakpoint was further verified using QPCR and Sanger Sequencing.Most of the CNVs were heterozygous or homozygous deletions, ranging from single-exon to whole-gene deletions.To detect CNVs, we utilized CNV-seq, a Next-generation sequence-based (NGSbased) CNV prediction with the gCNV algorithm[28].It requires an additional wet-lab assay to be applied to the samples that had already undergone NGS, which may be unnecessary if the CNVs can be detected by a robust NGS-based algorithm and is a cost-effective and easily accessible method widely.While CNV-seq was efficient in capturing large CNVs, it lacked precise localization of the breakpoint.Therefore, we turned to long-read sequencing as an alternative method to detect large CNVs.Long-read sequencing overcomes the limitations of the NGS.The third-generation technologies offer improvements in the characterization of genetic variation and directly detect the input molecule without DNA amplification or synthesis,therefore being mostly free from PCR-related bias.By employing ONT, we identified the copy number deletion region in chromosome 7q31.31-31.32 with the accurate breakpoint del(7)(q31.31q31.32)chr7:g.119451239_123956818del.
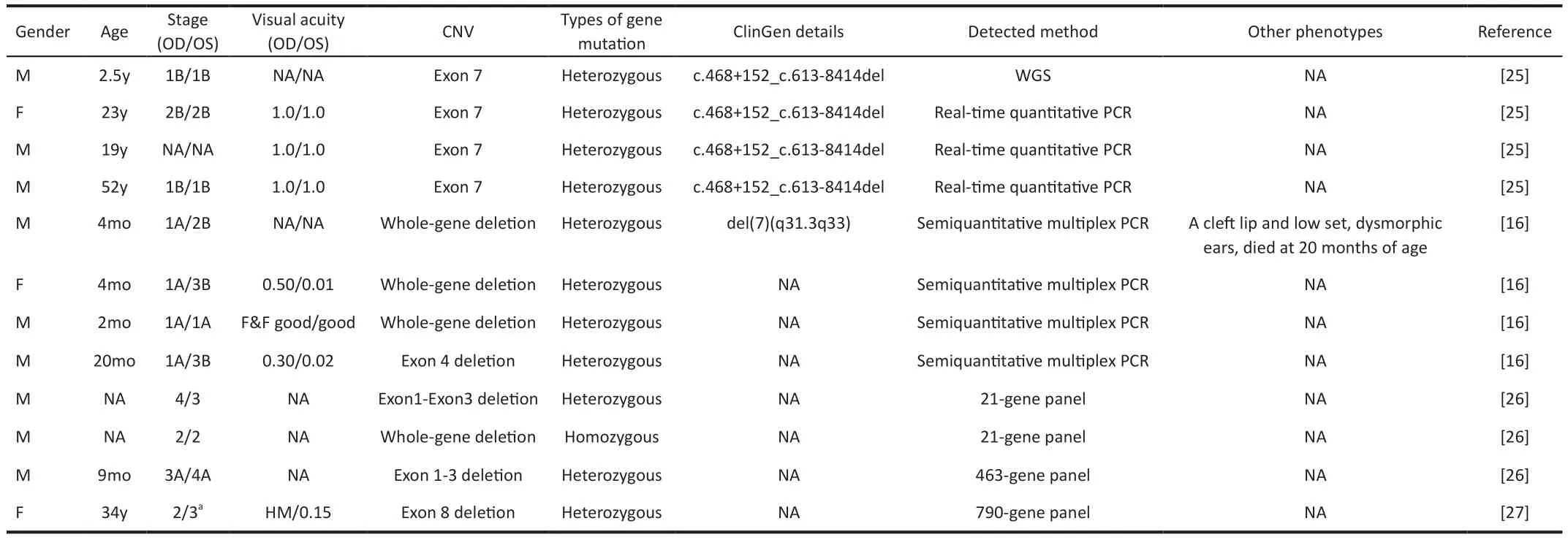
Table 4 Information about CNVs of TSPAN12 with FEVR phenotype
In conclusion, our findings suggest that the large deletion in chromosome 7q31.31-31.32, which includes theTSPAN12gene, is associated with an AD FEVR phenotype and moderate retinal vascular impairment.CNV is a genetic risk for FEVR patients, and genome sequencing provides an effective method to get the precise breakpoint of CNV, helping to identify genes associated with specific phenotypes.Further research is needed to investigate other dosage-sensitive genes associated with FEVR and elucidate their detailed functions in retinal angiogenesis.
ACKNOWLEDGEMENTS
The authors thank all patients and their family members for their participation.The authors also appreciate all clinicians for collecting of samples and clinical data.
Authors’ contributions:Sheng XL made the contribution to design the work, Zhang S write the manuscript, Yong HM, Ma MJ, Rui X and Yang SY collect the data, Zou G take part in analysis.
Foundations:Supported by the National Natural Science Foundation of China (No.82060183); Ningxia Natural Science Foundation (No.2022AAC03388); the Key Research and Development Project of Ningxia Hui Autonomous Region(No.2021BEG02045; No.2020BEG03044).
Conflicts of Interest: Zhang S,None;Yong HM,None;Zou G,None;Ma MJ,None;Rui X,None;Yang SY,None;Sheng XL,None.
杂志排行

International Journal of Ophthalmology的其它文章
- Dynamic tear meniscus parameters in complete blinking:insights into dry eye assessment
- Effects of diquafosol sodium in povidone iodine-induced dry eye model
- Morroniside ameliorates lipopolysaccharide-induced inflammatory damage in iris pigment epithelial cells through inhibition of TLR4/JAK2/STAT3 pathway
- Role of reactive oxygen species in epithelial-mesenchymal transition and apoptosis of human lens epithelial cells
- Electroacupuncture alleviates ciliary muscle cell apoptosis in lens-induced myopic guinea pigs through inhibiting the mitochondrial signaling pathway
- A pedigree with retinitis pigmentosa and its concomitant ophthalmic diseases