A pedigree with retinitis pigmentosa and its concomitant ophthalmic diseases
2023-12-14HongDouLuoShaoNanPeiAiJiaWangXueQingYuHaiJianHuLingZengFeiFeiWangMingJinXuZhang
Hong-Dou Luo, Shao-Nan Pei, Ai-Jia Wang, Xue-Qing Yu, Hai-Jian Hu, Ling Zeng,Fei-Fei Wang, Ming Jin, Xu Zhang
Affiliated Eye Hospital of Nanchang University, Jiangxi Research Institute of Ophthalmology & Visual Science; Jiangxi Provincial Key Laboratory for Ophthalmology, Nanchang 330006, Jiangxi Province, China
Abstract
● KEYWORDS: retinitis pigmentosa; glaucoma; wholeexome sequencing; RHO
INTRODUCTION
Retinitis pigmentosa (RP), the most common form of inherited retinal dystrophy (IRD), affects more than 1.5 million people worldwide.RP is characterized by the degeneration of photoreceptor cells and pigment epithelial cells and can lead to severe vision impairment or blindness[1].The typical clinical symptom at the initial stage is impaired dark vision due to the loss of rod photoreceptor cells[2], followed by progressive loss of the peripheral visual field.However,during the pathophysiologic process of RP, macular function is usually retained relatively well until the late stage of the disease.Fundus examination generally does not detect any abnormal symptoms until RP has progressed to an advanced stage at which the retinal fundus image shows retinal vascular stenosis, waxy color of the optic papilla and osteocyte-like pigmentation deposition in the periphery of the retina.
Primary nonsyndromic RP has significant genetic heterogeneity,and autosomal dominant retinitis pigmentosa (ADRP) is the most common type of hereditary RP, accounting for 20%-25%of cases[3].Mutations in 25 ADRP genes and an additional related gene,RP63, including more than 1000 dominant or nondominant mutations, have been reported to account for 50%-75% of ADRP[4].Among them, mutations in the rhodopsin (RHO) gene are the most common cause of ADRP worldwide.Since Dryjaet al[5]first discovered that a C>A transversion (Pro23His) in codon 23 ofRHOcan lead to RP in 1990, more than 200 point mutations inRHOhave been identified (https://sph.uth.edu/Retnet).
Vision loss in primary nonsyndromic RP is caused by gradual loss of photoreceptors and development of complications such as cystoid macular edema (CME), epiretinal membrane(ERM) and cataracts[6].Acute attack or chronic progression of glaucoma has also been shown to be a risk factor for rapid visual deterioration in patients with RP[7-8].Nevertheless, the causes of retinoschisis in some patients with RP need to be further explored.The purpose of this study was to detect the mutated gene in an ADRP family from Fujian Province, China, and to explore the relationship between gene mutation and clinical phenotype.
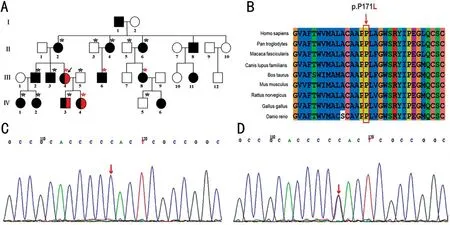
Figure 1 Pedigree data and gene mutation A: Pedigree data for an ADRP family.Pedigree of a large Chinese family with ADRP.Solid squares and circles denote affected males and females, respectively (black denotes RP, and red denotes suspected retinoschisis).Healthy individuals are represented by empty symbols.The proband is indicated by an arrow.Peripheral blood sampling for whole exon sequencing is indicated by a red asterisk.Peripheral blood sampling for Sanger sequencing is indicated by a black asterisk.B: Conservation analysis of the rhodopsin p.P171 amino acid residue, which is drawn by the software Clustalx.C: Sanger sequencing results of the normal people, II:3, II:5, III:5, IV:5.D:Sequencing results of the RHO gene showed a missense mutation from C to T at exon 2 [c.512C>T (p.P171L), red arrow] of II:2, II:4, II:6, III:2,III:3, IV:1, IV:2, IV:3.ADRP: Autosomal dominant retinitis pigmentosa; RP: Retinitis pigmentosa; RHO: Rhodopsin.
SUBJECTS AND METHODS
Ethical ApprovalThis research was carried out in strict accordance with the Helsinki Declaration.All subjects signed an informed consent form, and the research was approved by the Ethics Committee of the Ophthalmic Hospital affiliated with Nanchang University (No.202012011), Nanchang, China.ParticipantsA 30-person, four-generation Han-Chinese pedigree was recruited in Fujian Province in China.Sixteen patients with RP in this pedigree (among them, family members Ⅱ:8 and Ⅲ:11 are known to have RP based on their medical history inquiry).The pedigree was consistent with autosomal dominant inheritance (Figure 1A).The proband(Ⅲ:4) was diagnosed with binocular RP, binocular chronic angle-closure glaucoma, high myopia and cataracts at the Affiliated Eye Hospital of Nanchang University in November 2019.After acquiring the medical history of the proband,comprehensive clinical data were obtained from 8 affected members of the family (Ⅱ:2, Ⅱ:4, Ⅱ:6, Ⅲ:2, Ⅲ:3, Ⅲ:4, Ⅲ:6,Ⅲ:8).We performed whole exome sequencing (WES) on the proband (III:4) and three members of the family (III:6, III:8,IV:4).The selected candidate mutation sites were validated by Sanger sequencing on 12 members of the family (II:2, II:3,II:4, II:5, II:6, III:2, III:3, III:5, IV:1, IV:2, IV:3, IV:5).
Clinical ExaminationThe vision and intraocular pressure(IOP) of all participants were checked in the outpatient department.All participants underwent visual acuity testing by E decimal charts and optometry.The subjects also underwent detailed ocular examination, which included slit lamp biomicroscopy, best corrected visual acuity (BCVA),A-ultrasound, B-ultrasound, fundus photography, ultrasound biomicroscopy (UBM), optical coherence tomography (OCT)and electroretinography (ERG).
The diagnostic criteria of primary RP are as follows[9].1) A history of night blindness: unable to see or not clear in weak light or complete darkness, a lower ability to perceive weak light than that of normal people, and a longer adaptation time in darkness than that of healthy people.2) Visual acuity gradually decreases.3) Fundus manifestation: the retinal vascular wall was narrowed and thinned, and the optic disc was normal in the early stage.With the progression of the disease,the color of the optic disc can be white or waxy yellow.Needle-like or irregular pigmentation of patchy osteocytes appeared in the equatorial part or the middle periphery of the retina.In the late stage of the disease, atrophy of the retina and anterior scleral choroid can be seen in a wide range (or limited to a certain quadrant).4) Visual field changes: punctate or patchy loss of the peripheral visual field in the early stage and concentric narrowing in the late stage.5) ERG: the function of rod cells decreases in the early stage, and cone cells decrease slightly.The functions of cone and rod cells both decrease in the late stage.The amplitudes of the a wave and b wave decline abnormally and even disappear in the late stage.Primary RP can be diagnosed when patients meet two or more of the above diagnostic criteria, with exclusion of RP with other systemic abnormalities, syndromes and RP secondary to other diseases.
Molecular Genetic MethodsIn total, 3-5 mL peripheral venous blood samples anticoagulated with ethylenediamine tetraacetic acid (EDTA) were collected from all participants.DNA (≥20 ng/μL, QIAamp DNA Blood Midi Kit, Qiagen,Hilden, Germany) was extracted by the silica gel modeling technique and rapid centrifugation column operation.Genomic DNA with qualified purity was detected by ultraviolet spectrophotometry and agarose gel electrophoresis.WES was performed by MyGenostics Incorporation, Beijing, China,using gDNA from the proband (III:4) and three members of the family (III:6, III:8, IV:4).The cDNA database after PCR linear amplification, the database was checked and sequenced.The original imence by base recognition analysis, called RawData,RawData were finely filtered to obtain CleanData.CleanData were compared to the reference genome, and the sequencing depth and coverage of the target region were calculated.Then,CleanData were compared to the human genome by Burrows-WheelerAligner software.Single nucleotide variants (SNVs)and Insertions and deletions (InDels) were detected by GATK software and VarScan software, and multiple databases (mainly ExAC, dbSNP, 10 000 genome, GnomAD, InterPro, ClinVar,OMIM, HCMD,etc.) were used with ANNOVAR software to annotate the results.Before follow-up analysis, necessary emission sample filtering and pedigree analysis filtering were carried out on the variant data set.
Primers were designed for the mutant loci screened, PCR amplification was carried out, configuring the PCR amplification reaction system using the Beckman automated workstation preset program, and amplify using an A&B PCR instrument:98℃ for 2min; 98℃ for 10s, 65℃ for 30s, and 72℃ for 10s,a total of 10 cycles; 98℃ for 10s, 55℃ for 30s, and 72℃ for 10s, a total of 25 cycles; 72℃ 1min; cool and store at 4℃.The products were sequenced by Sanger sequencing.PCR amplification and Sanger sequencing primer sequences were as follows: 5’-AAGCTCTCTCCTTCCCCAAG-3’ and 5’-CCAGCCCTTGTAGCAACATT-3’.Cosegregation of mutant gene loci was verified in selected family members to determine the mutant gene loci that led to RP in the family.
RESULTS
Pedigree Clinical CharacteristicsThere were 16 individuals,including 6 males and 10 females, diagnosed with RP in this 30-person, four-generation pedigree.Of sixteen individuals diagnosed with RP, 8 (Ⅱ:2, Ⅱ:4, Ⅱ:6, Ⅲ:2, Ⅲ:3, Ⅲ:4, Ⅲ:6,Ⅲ:8) were included in the analysis.The affected individuals exhibited typical symptoms, including nyctalopia, progressive visual impairment, color vision deficiency and central visual field sensitivity loss, followed by progressive peripheral vision loss.The ophthalmic clinical data for eight patients are summarized in Table 1.The eye sight of the eight patients was poor, with an average visual acuity of 0.045±0.06 and BCVA of 0.22±0.15.The average axial length was 24.58±0.83 mm, which is slightly longer than that of healthy people.All eight patients had refractive errors.Residual central tubular field of vision was detected in the eight patients.No obvious abnormalities appeared in the macula and optic disc of the eight patients, but typical fundus changes, such as bone spicule-like pigmentation,were found.ERG showed significant abnormalities, even with no waveform.The probands (Ⅲ:4), Ⅱ:2, Ⅱ:4, Ⅲ:2, Ⅲ:3, andⅢ:6 had shallow anterior chambers to varying degrees, and their IOP appeared to be normal (Table 1).
The proband (Ⅲ:4) was a 32-year-old woman with biliary pain, vision loss and night blindness for two years.She was diagnosed with binocular RP, chronic angle-closure glaucoma,high myopia and complicated cataract.The proband developed bilateral vitreous opacity, and her BCVA was recorded as 0.15 in the right eye and 0.5 in the left eye.Fundus appearance showed a waxy yellow optic disc and attenuated retinal vessel, and bone spicule-like pigmentation could also be found in fundus photography (Figure 2A).Bilateral vitreous opacity appeared in both eyes.The visual field was defective in the peripheral area and displayed a tubular aspect (Figure 2D).ERG revealed an unrecordable scotopic and a photopic response (Figure 3).OCT showed atrophy in the outer layer of the retina and choroid and an elliptical area of choroidal vessels(Figure 2B).UBM examination revealed that the central depth of the anterior chamber became shallow.The iris was close to the anterior chamber angle and blocked the scleral process(Figure 2C), which is a sign of angle-closure glaucoma.
Whole-Exome Sequencing and Sanger SequencingTo identify pathogenic variants, WES was performed for the proband (Ⅲ:4) and three other family members (Ⅲ:6, Ⅲ:8,Ⅳ:4).The average sequencing depth of the target region was 103.98×, and a large proportion of the region, 98.7%, was covered by the target sequence at 20×.Possible candidate loci of mutation verified by Sanger sequencing were detected in the peripheral blood samples of 12 family members: 8 (Ⅱ:2, Ⅱ:4,Ⅱ:6, Ⅲ:2, Ⅲ:3, Ⅳ:1, Ⅳ:2, Ⅳ:3) were RP patients, but the other 4 (Ⅱ:3, Ⅱ:5, III:5, Ⅳ:5) were not diagnosed with RP.
The testing and verification results are shown in Figure 1C,1D.In the proband (III:4) and three other family members(III:6, III:8, IV:4) who underwent WES sequencing, one heterozygous variant, c.512C>T (p.P171L), in exon 2 of theRHOgene was suspected to be a pathogenic variant,whereby cosegregating gene mutation in the pedigree might lead to autosomal dominant/recessive RP type 4.Of the 12 participants for whom Sanger sequencing was performed,eight family members with RP were confirmed to carry this mutation (Ⅱ:2,Ⅱ:4, Ⅱ:6,Ⅲ:2, Ⅲ:3, Ⅳ:1, Ⅳ:2, Ⅳ:3), but the remaining four unaffected family members (Ⅱ:3, Ⅱ:5, Ⅲ:5,Ⅳ:5) did not harbor the relevant variant.The proline at position 171 (p.P171) ofRHOis highly conserved across different species (Figure 1B), indicating that the missense mutation at this site is pathogenic.The threedimensional structure of the rhodopsin protein is illustrated in Figure 4.SIFT analysis and PolyPhen-2 prediction suggest that the mutation is harmful.According to the criteria and guidelines for the classification of ACMG genetic variation,this variation conforms to the suspected pathogenic variation,which strongly demonstrates the pathogenicity of p.P171L variants.
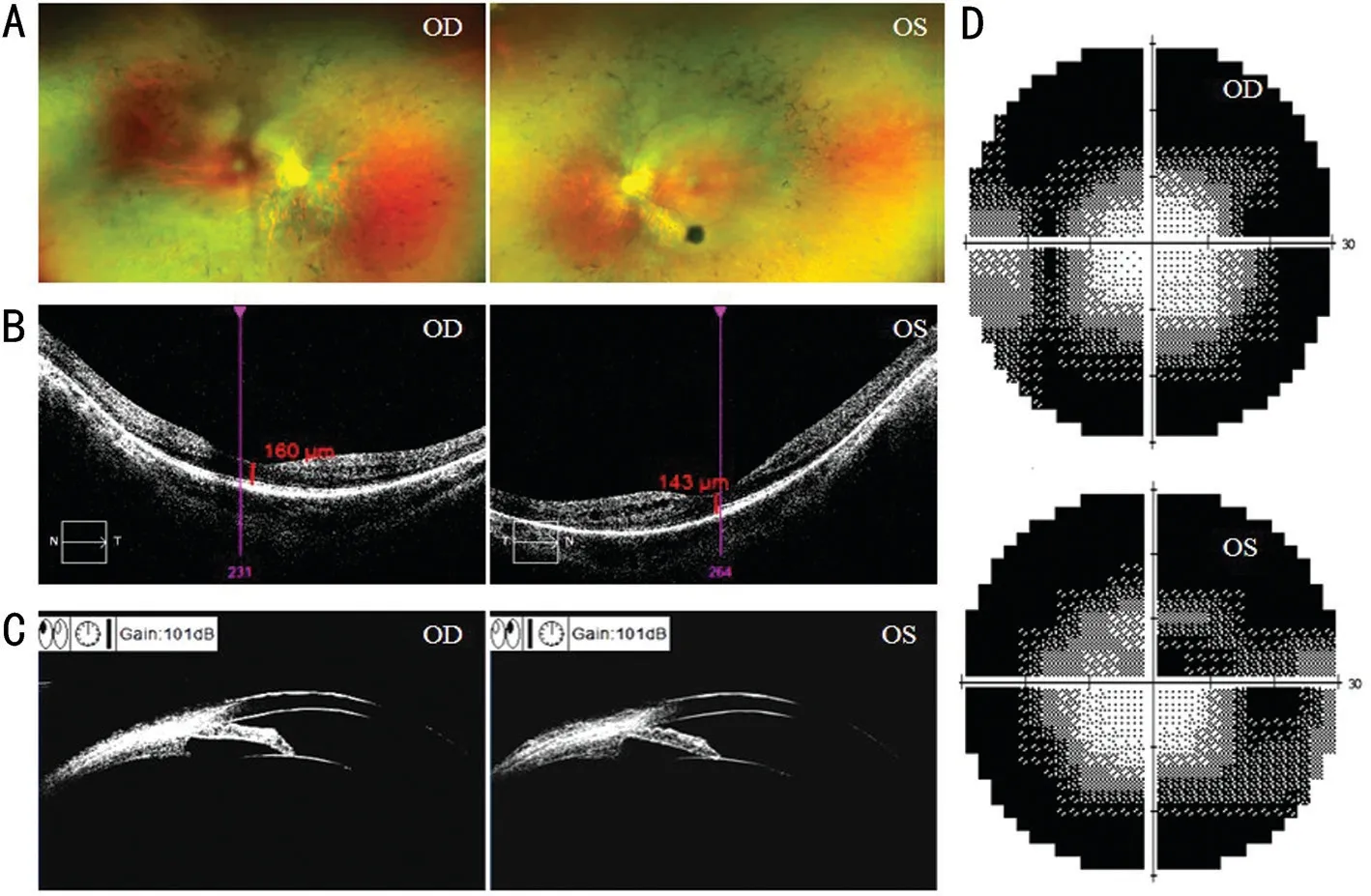
Figure 2 Fundus photography, OCT, UBM and perimetry of the proband A: Fundus photographs of the proband; B: Macular OCT of the proband; C: UBM of the proband; D: Perimetry of the proband.OD: Right eye; OS: Left eye; OCT: Optical coherence tomography; UBM:Ultrasound biomicroscopy.
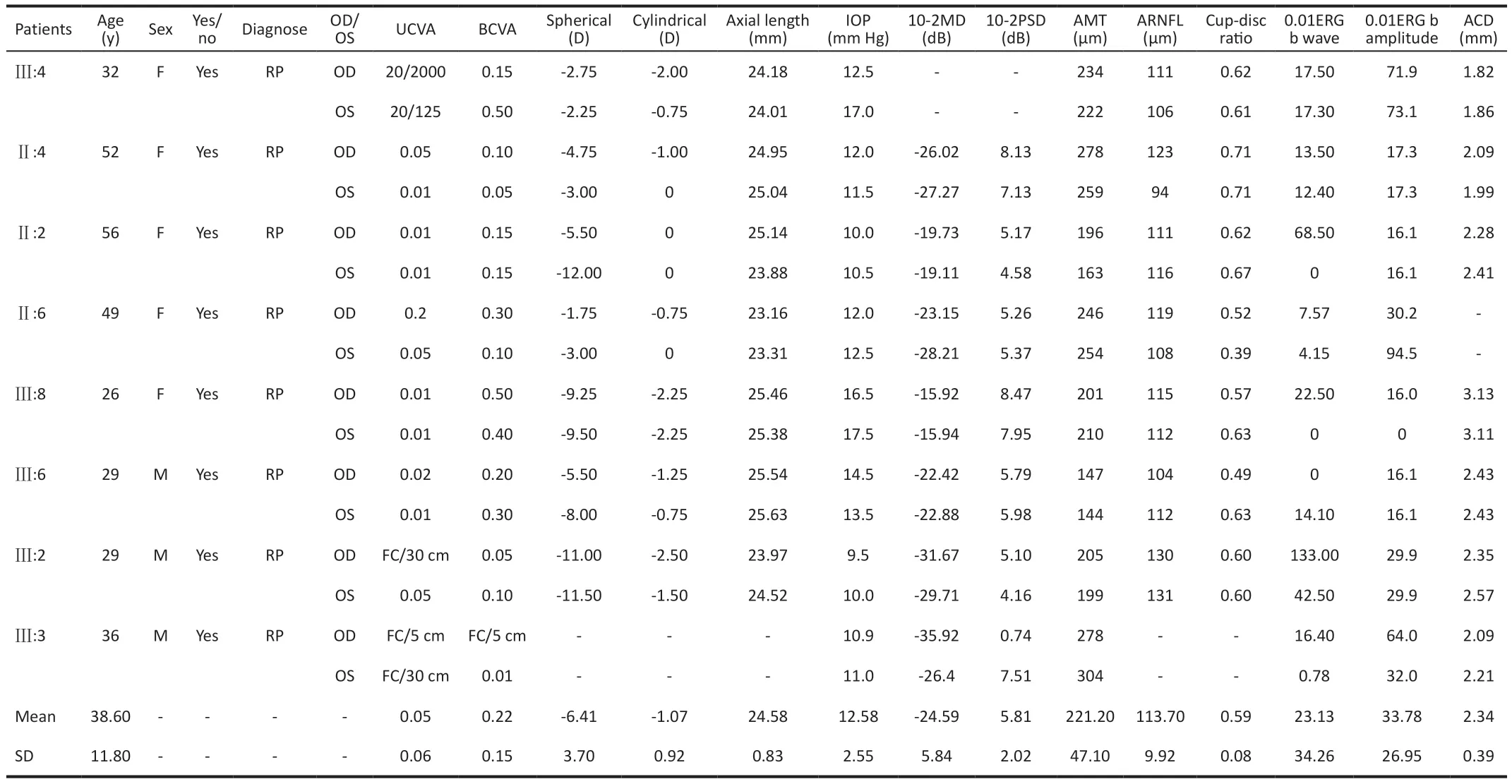
Table 1 Clinical data of eight pedigree members with the RHO c.512C>T (p.P171L) variant
DISCUSSION
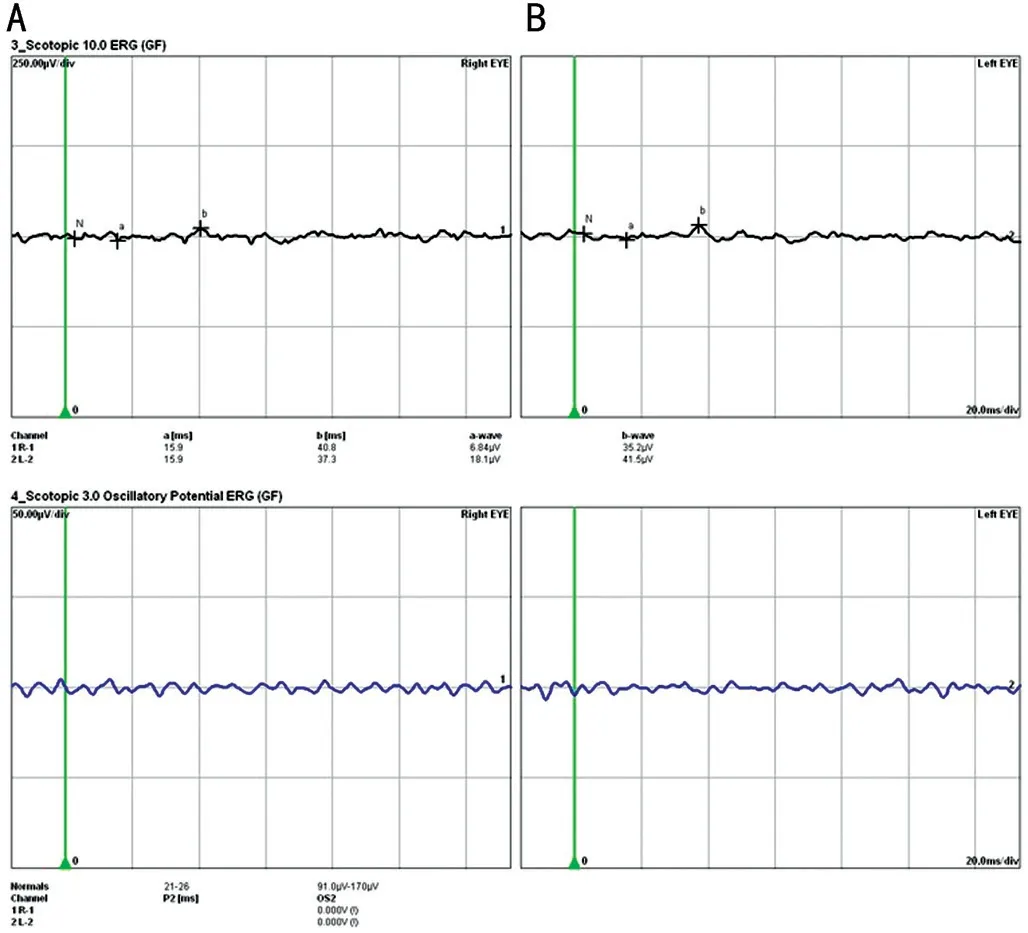
Figure 3 ERG of the proband Nonrecordable ERG results were observed in the proband’s right eye (A) and lefteye (B).ERG: Electroretinography.

Figure 4 Three-dimensional model structure of rhodopsin protein A: Overall structure of the protein; B: Amino acids, proline, at position 171.
Nathans and Hogness[10]first identified and sequenced the gene encoding human rhodopsin in 1984.TheRHOgene is located at chromosome 3q22.1 (build GRCh38.p10; http://www.ensembl.org/) with a span of 5 kb and encodes the rhodopsin protein composed of 348 amino acid residues[11].Rhodopsin is composed of opsin covalently linked to 11-cisretinaldehyde through Lys296[12].It is a G-protein-coupled receptor with seven transmembrane α helices and belongs to class A G-protein-coupled receptor[13].Rhodopsin has an outer segments targeting signal located in the cytoplasmic tail of the C-terminus, namely, the VXPX motif.Lack of the VXPX motif leads to dislocation of rhodopsin through mistransport and rod cell degeneration[14-15].The integrity of VXPX-COOH, a highly conserved outer segments targeting signal at the C-terminus of rhodopsin, is of great importance for photoreceptor cell survival,and mutation of VXPX-COOH can cause severe RP[16].
RP has a high degree of genetic and clinical heterogeneity,including autosomal recessive inheritance, autosomal dominant inheritance, X-linked recessive inheritance and double gene inheritance[3].RHOis the most studied RP gene,and the most common mutations are Ⅱ mutations.The Ⅱmutations inRHOare characterized by protein misfolding and can result in stress caused by retention in the endoplasmic reticulum.The P171L mutation inRHOis a class II mutation characterized by a missense mutation from C to T at exon 2 of the gene (512C>T).The codon changes from CCA to CTA, corresponding to a change from proline to leucine at the fourth transmembrane site of the rhodopsin protein (P171L)[17].Antiñoloet al[18]reported a Spanish family with the Pro171Qln mutation leading to ADRP.In this RP pedigree, the tertiary structure of opsin was destroyed due to codon 171 changing from CCA to CAA, and the patient showed onset of the disease at the age of 20 and developed a tubular visual field at the age of 40; the ERG waveform disappeared at the age of 30.
Taking the proband as an example, the patients with RP in this family were diagnosed with RP in infancy and exhibited relevant symptoms such as nyctalopia, visual acuity and progressive visual field reduction after 30 years of age.This is very similar to the clinical manifestations of a patient in an ADRP family with the P171L mutation reported by Wanget al[19], in which the presence of RP symptoms at an average age of 4-6y and night blindness appeared as the initial sign.After 20 years of age, the visual field gradually shrank, with even bilateral tubular central vision and visual impairment.Wanget al[19]speculated that the molecular mechanism of RP caused by the P171L mutation is that a nonpolar hydrophobic residue in the fourth transmembrane region of the rhodopsin protein changes the charge of residues in the lipid bilayer, resulting in instability.As proline is very important for the bending of peptide chains, the tertiary structure of mutant opsin may be disordered[17].In addition, the mutation is close to the binding site of the disulfide bond, which can cause the protein to be unstable.Mutations at the transmembrane site and around the disulfide bond usually cause a severe phenotype[20], which is consistent with the clinical phenotype observed for P171G[21].In this RP family, the proband (III:4) was diagnosed with chronic angle-closure glaucoma.II:2, II:4, III:2, III:3, and III:6 had shallow anterior chambers to varying degrees, and II:2 and II:4 had narrow anterior chamber angles to varying degrees.The proband exhibited high IOP and other typical glaucomatous pathological changes, such as iris bombe,shallow anterior chamber and long eye axis.However, the mild optic disc damage and the normal cup-disc ratio (C/D) were not consistent with the ocular hypertension of chronic angleclosure glaucoma.These clinical manifestations are similar to previous descriptions of patients with RP with glaucoma[7,22].The findings contribute to the phenotypic range of RP associated with glaucoma, which is helpful for clinical diagnosis in the future.Additionally, we analyzed the possibility of glaucoma susceptibility and family aggregation in this family.
Although clinical cases of RP combined with glaucoma are not common, the present study does show a correlation between these two diseases.In this study, we collected comprehensive clinical data from 8 RP patients and found that 12.5% had glaucoma and 75% had anatomical features of glaucoma.Koet al[23]conducted a retrospective analysis of 382 RP patients in Taiwan, China.The incidence of acute angle-closure glaucoma in RP patients was 3.64 times higher than that in normal controls.Wanget al[24]found that the prevalence of primary angle-closure glaucoma (PACG) in patients with RP was 2.88%, which was higher than the prevalence in the general population.Pradhanet al[25]concluded that the prevalence of PACG in RP patients over 40y was higher than that found in the general population of a similar age (3.8%vs0.8%).In our cohort of RP patients, 5.9% had primary angle-closure disease(PACD).The above data suggest a close relationship between RP and glaucoma.Acute attack or chronic development of glaucoma can lead to rapid deterioration of vision in RP patients, but glaucoma associated with RP is more likely to be missed in clinical diagnosis.The main reason is that there is no pain or other symptoms except in the acute attack period, and because RP will also lead to changes in vision, glaucoma is not easy to diagnose, which delays the best treatment.Therefore,it is of great significance to study the relationship between these two disorders, which will useful in clinical diagnosis and treatment as well as early intervention measures.
In addition, the triad with nanophthalmos, RP and angleclosure glaucoma as the main clinical manifestations has been reported[26-28], and this rare triad has been regarded as a recessive genetic syndrome.Furthermore, many studies have reported direct genetic evidence that RP-related gene mutations, such as inRPGRIP1[29],PRPF8[30], andRHO[8,28],are associated with glaucoma.TheRHOmutation found in this study provides a relevant basis for the genetic connection between RP and glaucoma.It is worth mentioning that in the clinical study of Wanget al[19]regarding the analysis ofRHOgene mutation in an RP family, the sameRHOmutation site,c.512C>T (p.P171L), was detected.Of particular interest,there were two RP patients with glaucoma in the study as well.Although there was a lack of further research, the similarities in clinical phenotype between these two studies indicate that P171L-mutantRHOis associated with glaucoma.There was only one proband diagnosed with glaucoma in our study; Ⅱ:2,Ⅱ:4, Ⅲ:2, Ⅲ:3, and Ⅲ:6 had anatomical characteristics of angle-closure glaucoma to different degrees, but glaucoma had not yet occurred.We presume that our findings support the view that RP-related gene mutations can increase glaucoma susceptibility.To prevent missed diagnosis, IOP measurements should be routinely carried out for RP patients, and the atrial angle should be checked for early diagnosis, early treatment and to delay blindness.In addition, if peripheral retinal examination cannot be performed with mydriasis for glaucoma, especially angle closure glaucoma, it is necessary to investigate whether the patient has night blindness and an RP family history to judge whether there is any impact on the visual field caused by RP.Once RP combined with glaucoma is diagnosed, it is recommended that the patient undergo a family survey and genetic counseling.
According to the results of clinical examination, all the patients in this family, including the proband, had varying degrees of myopia, and the BCVA of the proband, which was 0.15 in the right eye and 0.01 in the left eye, attracted our interest.The incidence rate of myopia among Chinese adults is approximately 50%, and some studies have shown that the incidence rate of high myopia is 0.55%[31].However, in the clinical data collected in this study, all patients had refractive errors, and the proportion of high myopia reached 42.86%.Myopia is one of the common ocular diseases associated with RP.Current studies show that moderate and high myopia can often be secondary to the RP phenotype in patients with RP or RP-related gene carriers[32-34].However, RP cases with high myopia are mostly associated with X-linked inheritance, and over 80% of X-linked RP cases are caused by RPGR gene mutations[34-35].Although it has been reported that there is a close relationship betweenRHOmutation-induced RP and myopia[36-37], the underlying mechanism between RP and myopia is not yet clear and must be investigated based on more similar clinical cases.On the other hand, Ⅲ:6 and Ⅳ:3 were diagnosed with cataracts in this pedigree.In previous studies,cataracts were recognized as the most common anterior segment complication of RP[38], and the prevalence rate of autosomal dominant genetic subtypes was higher than that of other genetic subtypes[39].Although the pathogenic mechanism of cataract associated with RP is not fully understood, Fujiwaraet al[40]noted that the Tyndall phenomenon is the key factor for cataract formation, and abnormal nutritional metabolism of the lens may be related to chronic inflammation.
In this pedigree, Ⅳ:3 and Ⅳ:4 were diagnosed as having retinoschisis associated with RP (Figure 5).In addition to typical RP symptoms such as progressive visual field damage and bone spicule-like pigmentation, they also had retinoschisis symptoms of abnormal splitting of the retinal layer and macular neuroepithelial bulging.Although the proband was not diagnosed with retinoschisis, she had thinning of the macular thickness, a common symptom found in patients with retinoschisis.According to the sequencing results of IV:3 and IV:4, no mutant genes related to congenital retinoschisis were found.Acquired retinoschisis mostly occurs in elderly individuals, which was not consistent with the age of the affected members in this pedigree.Therefore, we consider that there may be a certain concomitant relationship between RP and retinoschisis, and this finding has significance for clinical diagnosis and treatment.
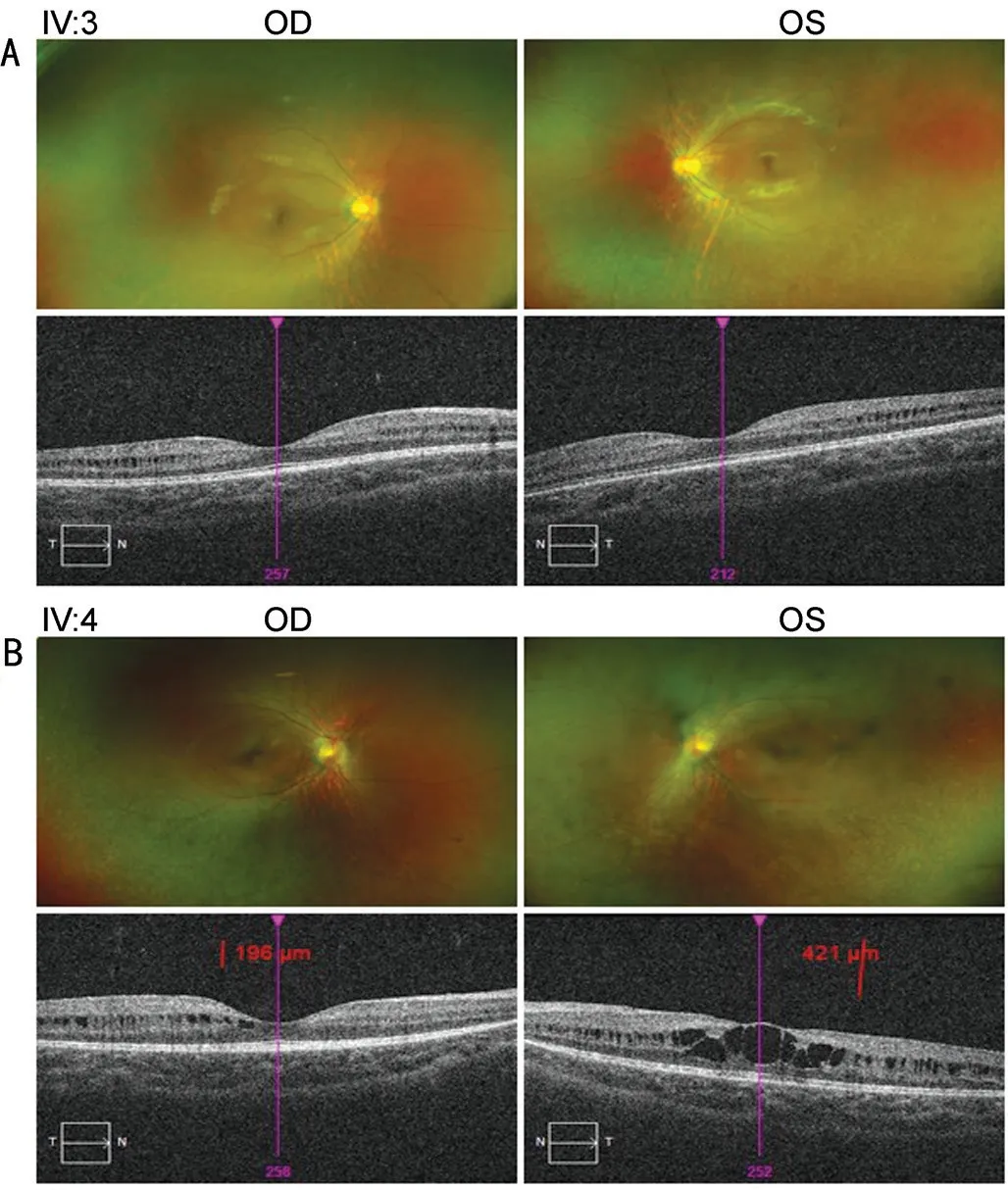
Figure 5 Fundus and OCT photographs of participants A: IV:3; B: IV:4.OCT: Optical coherence tomography; OD: Right eye; OS: Lefteye.
At present, the following are conjectures about the mechanism of RP associated with retinoschisis.1) The internal environment of rod cells and cones in RP patients changes during degeneration,which can result in abnormal expression, modification and secretion of RS1 protein and prevent it from performing adhesion functions, leading to retinoschisis.2) The connection between retinal layers of RP patients is weakened due to atrophy and degeneration.In this case, external traction caused by eye axis changes or introverted traction induced from the vitreous cortex, inner limiting membrane and retinal artery are likely to cause retinoschisis[41].3) Microglial reactivity exists widely in retinal degeneration or degenerative diseases such as RP and retinoschisis[42].Therefore, high levels of TNFα,MCP1, CCL2 and other factors in the outer nuclear layer of the retina lead to optic nerve cell apoptosis[43-44], resulting in thinning of the outer nuclear layer and weakening of the connection between the layers of the retina[45].Macular atrophy can also contribute to retinoschisis associated with RP to a certain extent[46-47].
As there are no reports and data supporting the concomitant relationship between RP and retinoschisis, our study on family members with RP and retinoschisis in this pedigree is important.The findings can broaden the phenotypic characteristics of the disease and indirectly suggest that there may be a certain degree of pathological relationship between RP and retinoschisis; however, the underlying mechanisms still need to be further explored.
In conclusion, in this study, we identified the missense mutation c.512C>T (p.P171 L) in exon 2 ofRHOresponsible for ADRP in a large Han Chinese family.We also discussed the relationship between RP and some ocular diseases in this pedigree, including glaucoma, myopia, retinoschisis,and cataracts.Our present findings support the existing view between RP and some ocular diseases and expand the genotype-phenotype relationship betweenRHOc.512C>T(p.P171L) and ADRP clinical findings and enrich the phenotypic range of RP-associated ocular diseases.The results will provide guidance for family genetic counseling and RP target gene therapy strategies in the future.
ACKNOWLEDGEMENTS
Foundations:Supported by the National Natural Science Foundation of China (No.81271425; No.81860170);the Natural Science Foundation of Jiangxi Province(No.20181ACG70010).
Conflicts of Interest: Luo HD,None;Pei SN,None;Wang AJ,None;Yu XQ,None;Hu HJ,None;Zeng L,None;Wang FF,None;Jin M,None;Zhang X,None.
杂志排行

International Journal of Ophthalmology的其它文章
- Dynamic tear meniscus parameters in complete blinking:insights into dry eye assessment
- Effects of diquafosol sodium in povidone iodine-induced dry eye model
- Morroniside ameliorates lipopolysaccharide-induced inflammatory damage in iris pigment epithelial cells through inhibition of TLR4/JAK2/STAT3 pathway
- Role of reactive oxygen species in epithelial-mesenchymal transition and apoptosis of human lens epithelial cells
- Electroacupuncture alleviates ciliary muscle cell apoptosis in lens-induced myopic guinea pigs through inhibiting the mitochondrial signaling pathway
- De novel heterozygous copy number deletion on 7q31.31-7q31.32 involving TSPAN12 gene with familial exudative vitreoretinopathy in a Chinese family