基于第一性原理分析铈对Al-Ti-B-Ce中间合金中TiB2 界面行为的影响
2023-12-05李龙泽傅高升陈鸿玲宋莉莉王火生
李龙泽 ,傅高升,2 ,陈鸿玲 ,宋莉莉 ,王火生
(1.福州大学机械工程及自动化学院,福州 350108;2.闽江学院物理与电子信息学院,福州 350108;3.福州大学材料科学与工程学院,福州 350108;4.福建工程学院材料科学与工程学院,福州 350108)
0 引言
Al-Ti-B中间合金细化剂主要用于铝及其合金的细化处理,以提高铝及其合金的综合力学性能。Al-Ti-B中间合金细化剂中的细化相主要为TiAl3相和TiB2相,其中TiB2细化相粒子因表面能较高,易发生聚集和沉降,形成密实型(堆垛聚集)的TiB2[1],这会导致细化剂的细化效果衰退[2]。作者所在课题组对稀土(RE)的细化、变质作用以及Al-Ti-B-RE中间合金细化剂的细化作用开展了大量的研究[3-7],发现在Al-Ti-B 中间合金中引入稀土铈后,不仅能够显著提升其细化效果,还能增强其细化长效性;进一步观察其SEM 形貌并进行能谱分析后发现,TiB2相更细小,分布更均匀,这是因为稀土元素作为表面活性物质,易在晶体表面和界面上吸附或偏聚,填补晶面上的缺陷,阻碍TiB2相的聚集。根据硼化物理论、超形核理论和双重形核理论[8-9],TiB2粒子或直接成为α-Al的异质形核核心,或成为TiAl3细化相的再生基底,因此TiB2的形态和分布状况是Al-Ti-B中间合金细化剂细化效果的重要影响因素[10-11]。从微观上看TiB2的堆垛聚集与TiB2的界面行为密切相关,铈对TiB2堆垛聚集的抑制作用也关系到铈对TiB2界面行为的影响,但是目前有关微观尺度下铈对TiB2界面行为影响的报道较少。基于此,作者使用基于密度泛函理论的第一性原理计算方法,通过对比TiB2的不同界面结构和热力学性质,筛选出密实型TiB2的稳定界面结构,计算了Al-Ti-B中间合金细化剂中掺杂稀土铈前后TiB2(0001)∥TiB2(0001)界面的黏附功以及铈在界面处的偏聚焓和在(0001)面的吸附能,分析了铈对TiB2界面行为的影响。
1 计算方法与界面模型的建立
1.1 计算方法
使用基于密度泛函理论的CASTEP 软件包[12],采用GGA-PBE泛函并结合超软赝势进行求解计算。最大平面波截断能设置为400 eV,作用在每个原子上的力不大于0.1 eV·nm-1,内应力不大于0.02 GPa,当前后2次自洽循环的每个原子能量差小于1×10-6eV时,即认为符合收敛条件。模型内的所有原子都具有3个方向上的自由度。布里渊区K点网格采用Monkhorst-Pack方法[13]划分,对于TiB2体相(指相对于表面或界面,晶体中具有理想周期性结构的主体),划分为10×10×8[14];对于所有表面和界面,划分为10×10×1,建立厚度为1.5 nm 的真空层以消除表面或界面间的相互作用,并在垂直表面或界面方向上添加偶极矫正以消除偶极效应[15]。为验证计算方法的可靠性,对TiB2体相进行结构优化,得到其平衡晶格常数,同时计算了其弹性模量、剪切模量和体积模量,并与他人的研究结果[16-18]进行对比。由表1可以看出,计算结果与他人研究结果一致,相对误差小于6.18%,验证了计算方法的可靠性。TiB2的晶体结构属于六方晶系,空间群为P63/mmm,其中钛原子和硼原子以ABAB顺序层状堆叠,硼原子位于钛原子围成的三棱柱中心[19]。
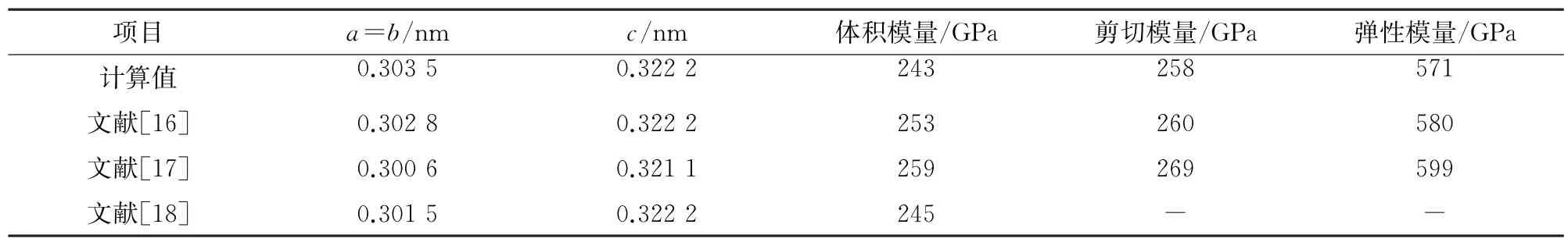
表1 计算得到TiB2 的平衡晶格常数、体积模量、剪切模量和弹性模量及他人研究结果Table 1 Calculated equilibrium lattice constant,bulk modulus,shear modulus and elastic modulus of TiB2 and study results by others
1.2 界面模型的建立
TiB2的各个晶面的生长速率按照从小到大的顺序依次为(0001)、{100}、{101}、{110}、{1-11},这种择优生长最终使得TiB2的(0001)面和(101-0)面外露,形成六角板片状的纳米晶[20]。纳米晶的表面能较高,倾向于通过聚集和长大来降低表面能。铝熔体中的六角板片状TiB2主要通过在(0001)面的平行堆垛以及连续横向铺展来聚集长大[21-22],因此选择TiB2的(0001)面作为研究对象。TiB2的(0001)面具有2种结构,分别是以钛原子为终端的表面结构和以硼原子为终端的表面结构,分别记为Ti-top 表面结构和B-top 表面结构。
合理的表面模型经充分弛豫后,除表面层原子外,其内层原子需要具有与体相内的原子相近的结构和性质,这需要TiB2表面模型具有足够多的原子层数[23]。TiB2表面模型中最内层原子层间距变化率ΔL表示为
式中:L'为表面模型最内层原子层间距;L0为体相原子层间距。
由图1可见,当TiB2(0001)表面的原子层数达到7层及以上时,其最内层原子层间距变化率已趋近于0,因此选择以7层原子的Ti-top表面和B-top表面建立TiB2(0001)∥TiB2(0001)界面模型。界面两侧原子(界面层原子)的堆垛方式会对界面的性质产生重要影响,而稳定界面的界面层原子堆垛方式一般为高对称性原子堆垛方式。高对称性原子堆垛方式可以分为上侧原子位于下侧原子正上方的顶位(top site)方式、上侧原子位于下侧相邻原子几何中心上方的心位(central site)方式、上侧原子位于下侧相邻原子连线中点的桥位(bridge site)方式[24]。作者考虑了8种最主要的高对称性原子堆垛方式形成的TiB2(0001)∥TiB2(0001)界面,使用VESTA软件[25]绘制的该界面在(0001)面垂直方向上的投影图如图2所示。为方便区分,根据组成界面的表面及其堆垛方式对这8种界面进行命名,将Ti-top和B-top表面分别记为T和B,顶位、心位和桥位方式分别记为TS,CS,BS,则TTTS代表Ti-top表面与Ti-top表面以顶位方式进行堆垛,形成的界面记为TTTS界面;其他7种界面命名以此类推。
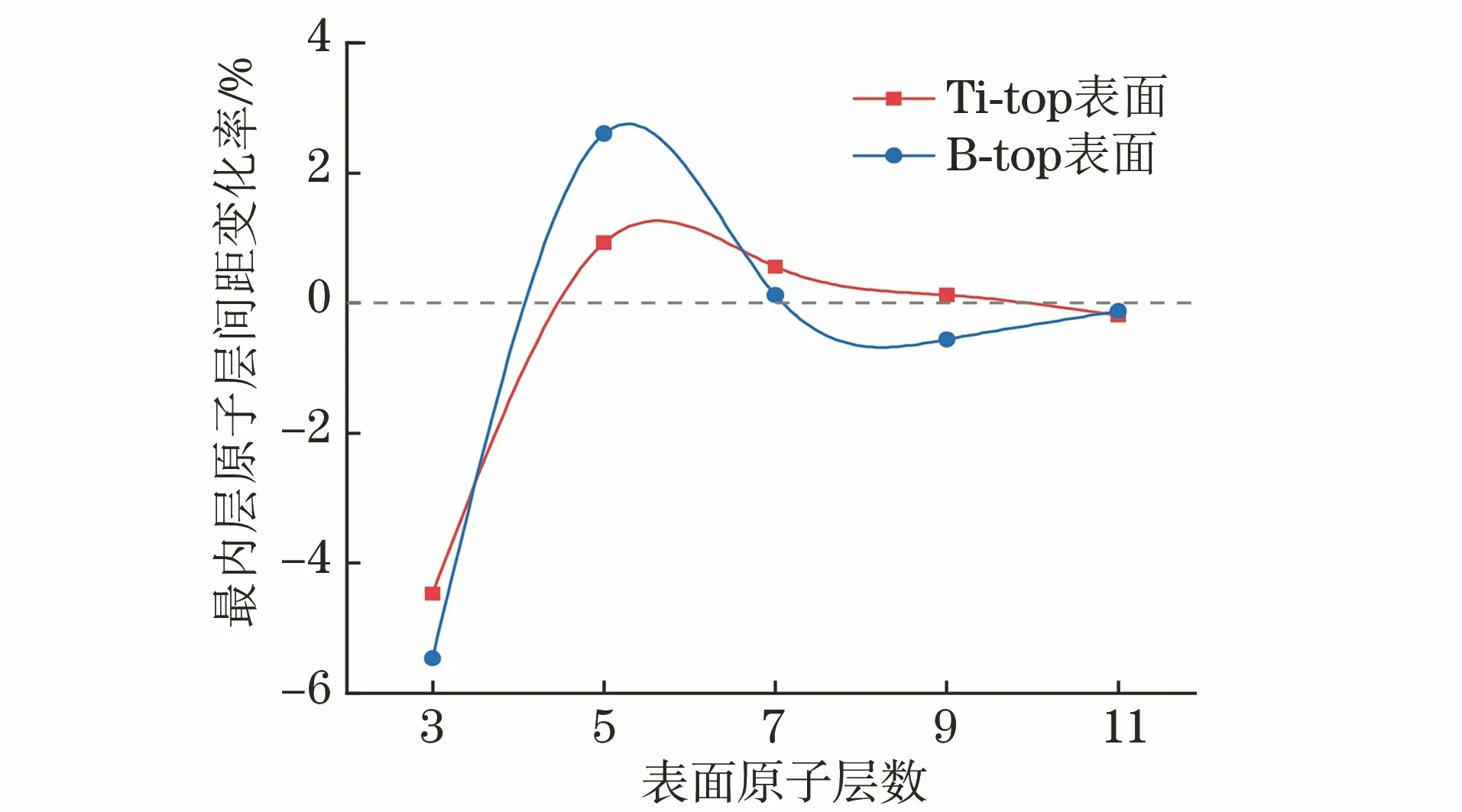
图1 TiB2(0001)表面模型最内层原子层间距变化率随原子层数的收敛趋势Fig.1 Convergence trend of innermost atomic layer spacing change rate with number of atomic layers of TiB2(0001)surface model

图2 8种高对称性原子堆垛方式形成的TiB2(0001)∥TiB2(0001)界面在(0001)面垂直方向上的投影图Fig.2 Projection of TiB2(0001)∥TiB2(0001)interface on(0001)plane along vetical direction formed by eight kinds of high symmetry atomic stacking methods
对8种TiB2(0001)∥TiB2(0001)界面模型进行充分弛豫,计算其黏附功和平衡界面距离以研究其热力学稳定性和结构稳定性。平衡界面距离表示在平衡状态下界面两侧原子之间的平均距离。黏附功(Wad)表示将凝聚相组成的界面Int分离成2个自由表面Slab1和Slab2时所需要的单位面积的可逆功,用来描述界面的结合强度。界面处黏附功(相界面结合能)[26]的计算公式为
式中:ESlab1和ESlab2为TiB2界面模型充分弛豫后分离为2个TiB2自由表面模型的能量;EInt为TiB2界面模型的总能量;A为界面面积。
黏附功越大,说明分离界面所需要的能量越高,TiB2在界面处的结合就越稳定;而平衡界面距离越小,说明界面两侧原子之间的相互作用越强[26]。通过对比8种TiB2(0001)∥TiB2(0001)界面模型的黏附功和平衡界面距离,筛选出相对稳定的界面结构。对筛选得到的相对稳定的TiB2(0001)∥TiB2(0001)界面掺杂稀土铈原子,研究稀土铈原子对TiB2(0001)∥TiB2(0001)界面的作用。通过计算偏聚焓Δseg来描述稀土铈在TiB2(0001)∥TiB2(0001)界面处的偏聚倾向,其本质为稀土铈偏聚到TiB2(0001)∥TiB2(0001)界面时体系能量的变化,具体表达式[27-28]为
式中:Eint·Ce为掺杂铈后界面模型的总能量;μCe为单个铈原子的能量;μx为被取代的单个钛原子或硼原子的能量。
当偏聚焓为负值时,偏聚焓的绝对值越大,偏聚过程自发进行的倾向越强;偏聚焓为正值表明偏聚过程需要吸收一定的能量才能进行,其值越小,所需要吸收的能量越少,偏聚过程也更易进行[28]。吸附能Ead能够反映稀土铈在TiB2(0001)表面上的吸附倾向,其本质为铈原子吸附在TiB2(0001)表面上使体系降低的能量。吸附能的计算公式[29]为
式中:Eslab为吸附铈前表面体系的能量;Eslab/Ce为吸附铈原子后表面体系的能量。
2 计算结果与讨论
2.1 黏附功和平衡界面距离
由图3可知:在由以钛原子为终端的2个Titop表面组成的TiB2界面(TTTS,TTCS,TTBS界面)中,以心位方式堆垛形成的TTCS界面的黏附功最高,平衡界面距离最短;在由以钛原子为终端的Ti-top表面和以硼原子为终端的B-top表面组成的TiB2界面(TBTS,TBCS界面)中,以心位方式堆垛形成的TBCS界面的黏附功最高,平衡界面距离与TiB2体相在垂直(0001)方向上的原子层间距0.161 1 nm 接近;在由以硼原子为终端的2个B-top表面组成的TiB2界面(BBTS,BBCS,BBBS界面)中,以桥位方式堆垛形成的BBBS界面的黏附功最高,平衡界面距离最短。根据自然界中普遍存在的能量最低原理,体系总是向最稳定、自由能最低的方向发展,同时在不同的钛环境下,TiB2的(0001)面倾向于以不同的原子为终端[15]。因此,从热力学和动力学的角度分析:当TiB2的(0001)面只存在Titop表面时,以(0001)面堆垛的密实型TiB2形成的界面主要为TTCS界面;当TiB2的(0001)面同时存在Ti-top和B-top表面时,以(0001)面堆垛的密实型TiB2形成的界面主要为TBCS界面;当TiB2的(0001)面只存在B-top表面时,以(0001)面堆垛的密实型TiB2形成的界面主要为BBBS界面。综上,在(0001)面上TiB2形成的相对稳定界面为TTCS界面、TBCS界面和BBBS界面,界面黏附功分别为4.11,8.32,4.54 J·m-2,均高于α-Al与TiB2的黏附功(3.18 J·m-2)[15],说明TiB2在(0001)面上具有较强的聚集生长倾向。
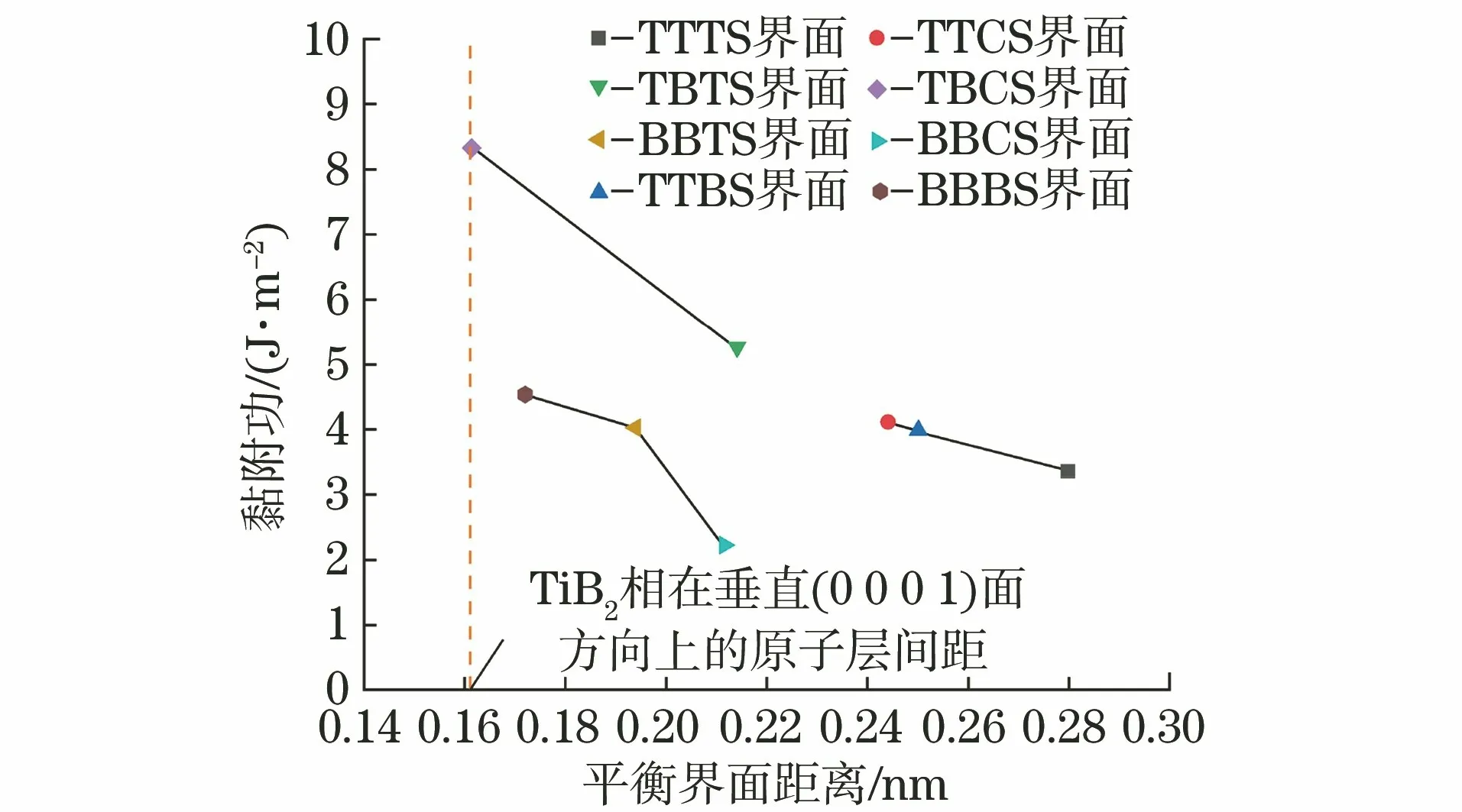
图3 8种高对称原子堆垛方式形成的TiB2(0001)∥TiB2(0001)界面的黏附功和平衡界面距离的关系Fig.3 Relation of adhesion work and equilibrium interface distance of TiB2(0001)∥TiB2(0001)interface formed by eight kinds of high symmetry atomic stacking methods
对TTCS界面、TBCS界面和BBBS界面这3种TiB2(0001)∥TiB2(0001)相对稳定的界面层中掺杂稀土铈原子,通过式(2)计算掺杂稀土铈原子前后TiB2(0001)∥TiB2(0001)相对稳定界面的黏附功。由图4可见:当在TiB2(0001)∥TiB2(0001)相对稳定界面处的钛位点掺杂稀土铈原子后,TTCS界面和TBCS界面的黏附功下降,表明分离界面所需的可逆功减小,TiB2的分散变得更为容易,在(0001)面上的聚集生长受到抑制;在TiB2(0001)∥TiB2(0001)界面处的硼位点掺杂稀土铈原子后,TBCS界面的黏附功下降,而BBBS界面的黏附功上升。从弛豫后的结构来看,取代硼原子的铈原子均脱离了原本硼原子所在的晶格位点,界面层原子发生了重排,平衡界面距离增大,表现出分离的趋势;由于铈的原子半径(0.224 9 nm)与硼的原子半径(0.0814 nm)相差较大[30],当硼原子被具有较大作用范围的铈原子取代后,铈原子周围产生较大的畸变,削弱了界面处原有键合的强度,使TBCS界面的黏附功下降;BBBS界面的黏附功在掺杂铈原子后略微上升,说明铈对B-top表面之间结合的抑制作用有限。在热力学上贫钛环境有利于B-top表面的出现,而在系统中存在足够的钛时,使TiB2以Titop表面存在的条件下,铈才会抑制所形成的TTCS和TBCS界面的结合[31]。

图4 掺杂铈原子前后TiB2(0001)∥TiB2(0001)相对稳定界面的黏附功Fig.4 Adhesion work of relatively stable interface of TiB2(0001)∥TiB2(0001)before and after doping with Ce atoms
2.2 偏聚焓和吸附能
由表2 可见,稀土铈在TiB2(0001)∥TiB2(0001)的3种相对稳定界面处的偏聚焓均为正值,这表明稀土铈在TiB2(0001)∥TiB2(0001)界面处的偏聚为吸热过程,不能自发进行。即使在偏聚焓相对较小的BBBS界面的硼位点,其偏聚焓也高达2.41 eV·atom-1,这说明当TiB2以(0001)面堆垛形成TiB2(0001)∥TiB2(0001)界面后,稀土铈难以对已形成密实型TiB2粒子在(0001)面的分散起到积极作用。

表2 稀土铈在TiB2(0001)∥TiB2(0001)相对稳定界面中的偏聚焓Table 2 Segregation enthalpy of rare earth Ce in relatively stable interface of TiB2(0001)∥TiB2(0001)
在Al-Ti-B合金细化剂的制备过程中引入稀土铈后,TiB2的聚集现象得到改善[4],或是因为稀土铈在TiB2(0001)表面上的吸附抑制了未形成密实型TiB2的粒子在(0001)面上的结合;但这需要稀土铈在TiB2(0001)面上具有吸附的热力学可能性。由表3可见,稀土铈在TiB2(0001)面的吸附能均为正值,且绝对值较高,表明稀土铈在TiB2(0001)面上具有较强的吸附倾向,其中在Ti-top表面上稀土铈的最稳定吸附位点为桥位,在B-top面上的最稳定吸附位点为心位。当稀土稳定吸附于TiB2表面时,能够有效降低以(0001)面形成的TiB2(0001)∥TiB2(0001)界面的黏附功,从而抑制TiB2在(0001)面的聚集生长。但当TiB2在(0001)面上形成界面后,由于稀土铈在TiB2(0001)∥TiB2(0001)上的偏聚焓为正值,稀土铈缺乏自发作用于界面的热力学条件,无法对已形成的密实型TiB2粒子起到分散作用,而是通过吸附于TiB2表面来抑制TiB2在(0001)面上的聚集生长。
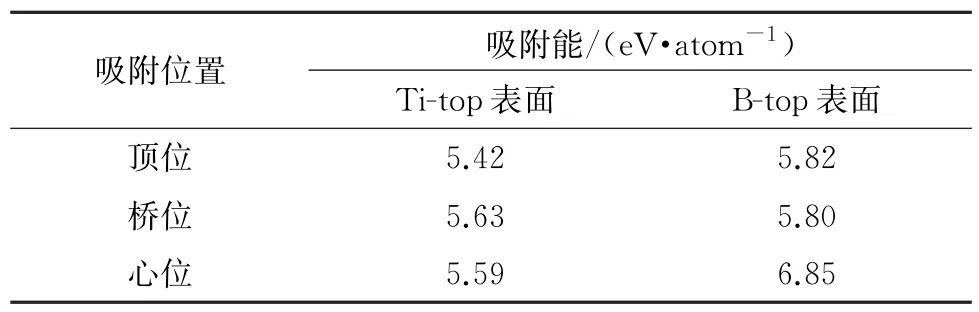
表3 稀土铈在TiB2(0001)面的吸附能Table 3 Adsorption energy of rare earth Ce on TiB2(0001)surface
2.3 局域电荷分布
局域电荷分布函数(ELF)对泡利不相容原理做出量化表述[32],表示在一个电子附近找到具有相同自旋电子的概率,是分析原子间电荷分布和化学键的有效方法[33]。为了进一步直观地研究TiB2(0001)∥TiB2(0001)界面处的电荷分布以及原子间的成键情况,对TiB2(0001)∥TiB2(0001)界面中的TTCS界面以及BBBS界面掺杂稀土铈前后的局域电荷分布进行了计算,结果如图5所示。由图5可以看出:未掺杂稀土铈时,TTCS界面区域的ELF值约为0.5,接近完全自由电子分布,表现为明显的金属键特征,TTCS界面两侧的钛原子之间形成了以金属键为主的Ti-Ti键;当稀土铈置换在TTCS界面处的钛原子后,原本界面处ELF值约为0.5的区域变小,且电子的离域程度增加,金属键减弱,TTCS界面的结合强度降低。未掺杂铈的BBBS界面两侧硼原子之间的ELF值在0.7~0.9,电子高度局域分布,界面两侧的硼原子之间形成了以共价键为主的B-B键,但相比于界面内部的B-B键,电子的局域程度较弱,成键强度也较弱,但仍能使BBBS界面保持较好的稳定性;掺杂稀土铈后,界面区域的电子局域分布区域消失,表明界面的B-B共价键消失,这有助于抑制界面的结合。
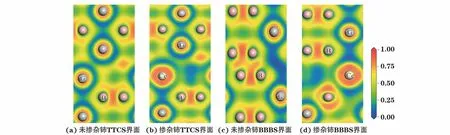
图5 未掺杂和掺杂稀土铈的TTCS界面和BBBS界面的局域电荷分布Fig.5 Electron localization function of TTCS interface(a-b)and BBBS interface(c-d)without(a,c)and with(b,d)Ce doping
综上可知,铈在TiB2(0001)∥TiB2(0001)界面处能够弱化界面两侧原子键合,使TiB2(0001)∥TiB2(0001)界面呈现出分离的趋势,也间接表明铈可吸附于TiB2表面对TiB2形成界面起到抑制作用。已有研究[34]表明,TiB2的(0001)面具有较大的形核潜力,可促进液相铝向固相α-Al转变。铈能减轻TiB2在(0001)面的聚集,为α-Al的形核提供更多的形核基底,并可作为Al-Ti-B-RE中间合金细化剂中TiAl3/Ti2Al20RE壳层结构相的异质形核核心[35],这有利于提高细化剂的细化性能,强化铝及其合金细化后的综合性能。
3 结论
(1) 在8种高对称性原子堆垛方式形成的TiB2(0001)∥TiB2(0001)界面中,具有稳定热力学性质和结构的界面为TTCS界面、TBCS界面和BBBS界面,且界面黏附功均高于α-Al与TiB2的黏附功,TiB2在(0001)面上有较强的聚集生长倾向。
(2) 掺杂稀土铈后TiB2(0001)∥TiB2(0001)相对稳定界面中TTCS、TBCS界面的黏附功降低,平衡界面距离增大,界面的结合强度降低,TiB2在(0001)面上的堆垛生长倾向降低。
(3) 稀土铈在TiB2(0001)∥TiB2(0001)相对稳定界面中具有正的偏聚焓,不能自发偏聚到界面处,无法促进已在(0001)面上聚集成团的TiB2分散;但铈在TiB2(0001)表面上具有较高的吸附能,能够稳定吸附在TiB2(0001)表面上,抑制TiB2粒子在(0001)面上的堆垛生长。
(4) 稀土铈削弱了TiB2(0001)∥TiB2(0001)界面两侧的原子键合,增加了TiB2表面形成界面的难度,有助于增加TiB2(0001)面形核基底的数量,更好地发挥Al-Ti-B-Ce中间合金细化剂的细化潜力。
