Vagus nerve stimulation protects against cerebral injury after cardiopulmonary resuscitation by inhibiting inflammation through the TLR4/NF-κB and α7nAChR/JAK2 signaling pathways
2023-11-27ShuangXuLangGuoWeijingShaoLicaiLiangTingtingShuYuhanZhangHeHuangGuangqiGuoQingZhangPengSun
Shuang Xu, Lang Guo, Weijing Shao, Licai Liang, Tingting Shu, Yuhan Zhang, He Huang, Guangqi Guo,Qing Zhang, Peng Sun
1Department of Emergency Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China
2Department of Urology, the Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou 510000, China
3Department of Emergency Medicine, the Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou 310009, China
4Department of Intensive Care Unit, Wuhan Hospital of Traditional Chinese Medicine, Wuhan 430000, China
5Department of Intensive Care Unit, Xiangyang Central Hospital, Affiliated Hospital of Hubei University of Arts and Science, Xiangyang 441021, China
6Department of Emergency, General Hospital of Central Theatre Command of the Chinese People’s Liberation Army,Wuhan 430070, China
7Department of Anesthesiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology,Wuhan 430022, China
KEYWORDS: Cardiopulmonary resuscitation; Vagus nerve stimulation; Inflammation; Toll-like receptor 4; α7 nicotinic acetylcholine receptor
INTRODUCTION
Cerebral injury after systemic ischemia-reperfusion(I/R) is a crucial cause of mortality and morbidity after cardiac arrest/cardiopulmonary resuscitation (CA/CPR).[1]The excessive inflammatory response is critical for the occurrence of nerve functional damage after CA.[2,3]Our previous studies showed that toll-like receptor 4 (TLR4)- mediated inflammatory immune damage plays an important role in this process.[4,5]Therefore, inhibiting inflammation induced by I/R is the key to alleviating ischemic brain damage and improving clinical outcomes after CPR.[6]The cholinergic anti-inflammatory pathway(CAP) is a feedback mechanism of endogenous neurons,[7]which regulates the excessive inflammatory response by inhibiting the activation of a downstream inflammatory signaling pathway, and is mediated by the activity of nicotinic acetylcholine receptor alpha 7(α7nAChR).[8-10]
α7nAChR is related to synaptic plasticity,neuroprotection, and neuron survival.[11,12]In our previous studies, we established a rat model of CA to prove that vagus nerve stimulation (VNS) can inhibit the inflammatory cytokines after CPR by activating α7nAChR-mediated CAP and improve the prognosis of resuscitation.[13,14]However, this still needs to be verified in more models.
Kim et al[15]have found that nicotine has a protective effect against endotoxemia by activating α7nAChRmediated CAP and thereby inhibiting TLR4 expression.It has been proven that the whole-body I/R injury caused by CA/CPR can be alleviated by VNS via α7nAChRmediated CAP.[14]However, similar to a model of endotoxemia, whether there is a crosstalk between the VNS-activated α7nAChR signaling pathway and the CPR-activated TLR4 signaling pathway in a model of CA/CPR is still unknown.
In this experiment, a mouse model of CA/CPR was established to verify the anti-inflammatory and protective effects of VNS.Meanwhile, the role of activated CAP in CPR and its molecular mechanisms were further explored by using the oxygen/glucose deprivation/reoxygenation(OGD/R) modelin vitro.
METHODS
Experimental animals and cells
Male C57BL/6 mice (8–12 weeks, 20–25 g)were obtained from the Experimental Animal Center at Wuhan, China.All experimental protocols of this animal experiment were carried out in accordance with the Animal Care and Use Guidelines of the Ministry of Science and Technology of the People’s Republic of China and were approved by the Medical Ethics Committee of Huazhong University of Science and Technology (Permit Number: S2356).
BV-2 cells were obtained from the Cell Resource Center of Peking Union Medical College (Beijing,China), and were cultured in high glucose DMEM(Hyclone) supplemented with 10% fetal bovine serum(FBS) (Sciencell, USA) and antibiotics 100 U/mL penicillin and 100 mg/mL streptomycin (Servicebio,China) at 37 ˚C in a 5% CO2incubator in saturated humidity.
Establishment of animal models of CA/CPR
The operation to induce CA is consistent with our previous experiments.[4,16]In brief, precordial chest compression, together with mechanical ventilation, was initiated after 8 min of CA induced by the injection of potassium chloride.At 8 min, delivered oxygen was adjusted to 100%, and mechanical ventilation was restored at a tidal volume of 0.7 mL and a breathing rate of 105 breaths/min (Ventilator: ALC-V8S, Shanghai Alcott Biotechnology Co., Ltd., China).Chest compressions were performed with a single finger at 300 stroke/min until sinus rhythm of electrocardiograph (ECG) appeared.[17]If the rescue time exceeded 10 min, the mice could not return to spontaneous circulation (ROSC), and the subsequent resuscitation was terminated.When spontaneous ventilation was sufficient, the ventilator was disconnected and the trachea was extubated.
To observe the neuroprotection of VNS after CPRin vivo, we randomly divided the mice into five groups.The sham group was intubated only in the right jugular vein(n=6).In the CPR group, the animals underwent 8 min CA followed by CPR (n=12).In the VNS group (n=12),except those mice were exposed to VNS 5 min before CPR, the treatment of mice was identical to that of the CPR group.The GTS-21 (α7nAChR agonist) group(n=12) was the same as the CPR group, except that GTS-21 (4 mg/kg) was injected 30 min before CPR.In the MLA (methyllycaconitine citrate, inhibitor of α7nAChR)+VNS group (n=12), the treatment was the same as in the VNS group, except that MLA (4 mg/kg) was given 30 min before CPR.GTS-21 was dissolved in saline and administered intraperitoneally in mice at a dose of 4 mg/kg,[18]beginning 30 min before CA/CPR.In a subset of animals, MLA (Tocris, Product No.1029, USA) was dissolved in DMSO and administered intraperitoneally at a dose of 5 mg/kg, beginning 15 min before VNS.[19]
Vagus nerve stimulation
The left cervical vagus nerve was separated through the median cervical incision and placed on a bipolar silver wire electrode for stimulation (Chengdu Taimeng Software Co., LTD., China).Electrical stimulation(square wave; intensity 1 mA; frequency 10 Hz; duration 1 ms) was applied for 5 min.[20]In the sham group, the vagus nerve was exposed but not stimulated.After the operation, the mice received buprenorphine (0.15 mg/kg)as an analgesic.
Establishment of the OGD/R model and drug administration
The OGD/R injury model is frequently used in the cerebral ischemia research.The cells were placed in a hypoxia tri-gas incubator (Thermo, USA) filled with N2, CO2, and O2(94%, 5%, and 1%, respectively) and D-hank’s for 3 h to achieve hypoxia.Next, the cells exposed to ischemia were returned to the normoxic incubator containing 95% air and 5% CO2, and cultured in DMEM with 10% FBS for another 24 h.The cells were seeded into 24-well plates (105cells/well for ELISA, 104cells/well for laser scanning confocal microscope), and 6-well plates (1.2×106cells/well for western blotting).
To explore the mechanism of VNS and to assess the effect of α7nAChR after I/Rin vitro, we used nicotine to activate the α7nAChR on BV-2 cell surface under OGD/R conditions, which mimic the I/R injuryin vitro.A concentration of 1 µmol/L nicotine was used in the subsequent experiments or vehicles.BV-2 cells were allocated into seven experimental groups: (1) normal culture (control, there was no OGD/R for cells) group;(2) nicotine (Sigma-Aldrich, USA) group (the cells were cultured in a standard incubator [95% air and 5% CO2]containing 1 μmol/L nicotine without OGD/R); (3) I/R group (OGD/R processing); (4) I/R+nicotine group (the cells were cultivated in a complete medium with 1 μmol/L nicotine before OGD/R); (5) I/R+nicotine+MLA group(before adding 1 μmol/L nicotine, the cells were cocultured in 10 μmol/L MLA for 30 min, and then OGD/R was performed); (6) I/R+nicotine+AG490 [inhibitor of Janus kinase 2 (JAK2)] (Product No.S1143 Selleck,China) group (before adding 1 μmol/L nicotine, the cells were co-cultured in 10 μmol/L AG490 for 30 min,and then OGD/R was performed); and (7) I/R+PDTC(inhibitor of NF-κB, Product No.s1808, Beyotime,China) group (before adding 1 μmol/L nicotine, the cells were co-cultured in 50 μmol/L PDTC for 30 min, and then OGD/R was performed).
Evaluation of neurologic deficit score (NDS)and survival rate
NDS assessment was performed for 3 consecutive days, including an assessment of consciousness, brain reflex, corneal reflex, breathing, auditory reflex, righting reflex, and motor function (0–500 scale; 0 normal, 500 death or brain death).[21]The 72-hour survival rate of mice in each group was analyzed.
Cell viability assay
Cell viability was proved with the MTT method.A spectrophotometer was used to measure the optical density (OD) values of 100 μL equivalents at 570 nm(Microplate Reader, Thermo Scientific™, USA).
Enzyme-linked immunosorbent assay (ELlSA)
According to the manufacturer’s instructions, ELISA kits were used to detect inflammatory factors in tissue or cell samples, such as tumor necrosis factor-α (TNF-α)(Biomart Technology, China) and interleukin- 6 (IL-6).
Laser scanning confocal microscope
TLR4 in BV-2 microglial cells was located and analyzed by laser scanning confocal microscope.BV-2 microglias (104cells/mL) were plated on coverslips in 24-well culture plates.After treatment, the BV-2 microglial cells were washed twice with PBS, fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked with 2% BSA for 1 h.Then, the BV-2 microglial cells were incubated with TLR4 antibody (1:100) at 4 ℃ overnight,and incubated with a FITC-conjugated secondary antibody (1:100) at 37 ℃ for 1 h, and then stained with DAPI.A laser scanning confocal microscope was used to obtain fluorescent images.
Western blotting
Total protein was extracted from cells or brain samples with RIPA lysis buffer (Beyotime Biotech,China), and centrifuged at 12,000 r/min for 10 min at 4 ℃.The supernatant was quantified with a BCA protein assay kit (Beyotime Biotech, China).An equal amount of protein samples (30 μg) was separated by 10% SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA).The membrane was sealed with 5% skim milk powder in Tris-buffered saline containing 0.05% Tween-20 (TBST)for 1 h, incubated with a primary antibody against α7nAChR (1:1,000; ab10096; Abcam), TLR4 (1:1,000;ab13556; Abcam), NF-κB p65 (1:1,000; 8242S; CST),phosphorylated (p)-NF-κB p65(1:1,000; 3033S; CST),JAK2 (1:1,000; 3230S; CST), phosphorylated (p)-JAK2(1:1,000; 3776S; CST), and GAPDH (1:500; Servicebio,China) overnight at 4 ˚C and washed with TBST (3×15 min).The corresponding horseradish peroxidase-(HRP-) conjugated secondary antibody (EarthOx, LLC San Francisco, USA) was added and incubated for 1 h at room temperature, followed by washing with TBST (3×10 min).Bands were visualized by an ECL chemiluminescence kit (K-12045-D10, Advansta,USA) and images were obtained using a Gel Doc™ EZ System (Bio-Rad Laboratories, Inc, USA).Image Lab™software (version 6.0, Bio-Rad Laboratories, Inc.) was used for density analysis.
Histopathological analysis
Three days after ROSC, the mice were deeply anesthetized and perfused through the heart with 4 ℃PBS.The mice were then decapitated and their brains were exposed to 4% paraformaldehyde overnight.Then,the sections were dehydrated with 70%, 80%, 90%,95%, 100% ethanol, transparent with xylene, embedded,sectioned, and detected with hematoxylin-eosin (HE)staining.For stained images, a light microscope(Olympus BX51, Japan) with a magnification of 40× and 400× was used.Our observations focused on the CA1 area of the hippocampus, which is considered the most sensitive area to hypoxia and ischemia.
Statistical analysis
All data were analyzed by SPSS 22.0 (IBM, USA)and expressed as mean ± standard deviation (SD).The comparison between groups was made by using ANOVA with Tukey’s post-test analysis or the Student’st-test(two-tailed).All statistical analyses were performed using GraphPad Prism 7.0.APvalue < 0.05 was considered to be statistically significant.
RESULTS
The baseline characteristics of the mice
There were no significant differences in body weight,temperature, heart rate or CPR times among the groups(Supplementary Table 1).
VNS improved 72-hour survival and neurological recovery in mice after resuscitation
As shown in Figure 1A-B, after observation for 72 h,compared with the CPR group, the VNS group and GTS-21 group had much higher survival rates (P<0.05).There was no significant difference in survival among the VNS group, GTS-21 group, and MLA +VNS group.
The NDS of each mouse was measured at 24 h, 48 h,and 72 h after ROSC.The CPR group and MLA +VNS group developed severe neurological deficits at 24 h, 48 h, and 72 h after ROSC (P<0.05).There seemed to be less neurological dysfunction in mice in the VNS group and GTS-21 group than in the CPR group (Figure 1 C-E).
VNS reduced damage to hippocampal neurons from CA/CPR
As shown in Figure 2A, after CA/CPR, different degrees of neuronal damage occurred in the CA1 area of the mouse hippocampus, such as severe nuclear pyknosis, nuclear fragmentation, neuronal edema, and tissue structure disorder.However, in the VNS and GTS-21 groups, the above-mentioned changes in neurons were less severe than those in the CPR group.Nevertheless,VNS-induced neuroprotection was reversed by MLA treatment.
VNS inhibited inflammatory reactions in mice after CA/CPR
To investigate whether VNS treatment may induce anti-inflammatory effects, we examined the level of IL-6 and TNF-α in the serum of mice after CA/CPR.As shown in Figure 2B-C, the levels of IL-6 and TNF-α were significantly increased in CPR group compared to the sham group (P<0.001,P<0.05).Compared with the CPR group, the above indicators of the VNS group and the GTS-21 group were significantly lower (P<0.05).The concentrations of inflammatory mediators in the GTS-21 group and the VNS group were similar.However, MLA treatment significantly reversed the anti-inflammatory effect (bothP<0.05).The results demonstrated that VNS improved neuroinflammation in CA by activating the expression of α7nAChR.
l/R induced the release of pro-inflammatory cytokines in microglia
After the BV2 cells were exposed to ischemia conditions for 1 h, 2 h, 3 h, 4 h, and 5 h respectively,they were placed back into a normal oxygen incubator for another 24 h.The culture medium was collected,and the levels of the inflammatory cytokine TNF-α were measured using ELISA kits.TNF-α peaked after 3 h of ischemia treatment following 24 h of reperfusion,we chose 3 h as the ischemia time for subsequent experiments (Supplementary Figure 1A).
Nicotine attenuated the release of TNF-α and MLA reversed this effect
BV-2 cells were incubated with nicotine at different concentrations in the initial experiments.As shown in Supplementary Figure 1B-C, 1 μmol/L–105μmol/L nicotine did not affect BV2 cell viability compared with the untreated group and 1 μmol/L nicotine obviously down-regulated the level of TNF-α.We used 1 μmol/L nicotine for subsequent experiments.Compared with the control group, I/R induced an approximately 3-fold increase in TNF-α expression (P<0.05).Treatment with 1 μmol/L nicotine significantly decreased the expression of TNF-α (P<0.01).To address the pivotal role of α7nAChR in mediating the anti-inflammatory effects of nicotine in microglia, we added MLA and AG490 to the I/R + nicotine group, and we found that nicotine failed to reduce TNF-α release by I/R in the presence of MLA or AG490 (Figure 3), indicating that nicotine exerts protective effects via α7nAChR/JAK2 signaling in microglia.
Nicotine inhibited the TLR4/NF-κB pathway by activating α7nAChR/JAK2 axis in BV-2 cells after l/R
As shown in Figure 4A and 4F, TLR4 was highly expressed in BV2 cells after I/R treatment and nicotine reduced its expression.However, MLA or AG490 treatment reversed this effect of nicotine obviously.Nicotine treatment not only prevented the expression of TLR4 and phosphorylation of NF-κB p65 in response to I/R in microglia, but also promoted the expression of α7nAChR compared with that in the I/R group.Conversely, the treatment with PDTC or MLA abolished these changes (Figure 4A and 4B).In I/R+nicotine group, we also found that the expression of p-JAK2 was increased in line with the expression of α7nAChR compared with that in I/R group, whereas, the addition of MLA or AG490 diminished this effect (P<0.05) (Figure 4C-E).Altogether, these results suggest that nicotine could upregulate the expression of p-JAK2 via the activation of α7nAChR, and downregulate the expression of TLR4 and p-NF-κB p65 in microglia after I/R.These results verified the assumption that the α7nAChR/JAK2 axis could participate in the anti-inflammatory effect of nicotine and TLR4/NF-κB might be negatively regulated by activating a7nAChR/JAK2 axis in BV-2 cells after I/R.
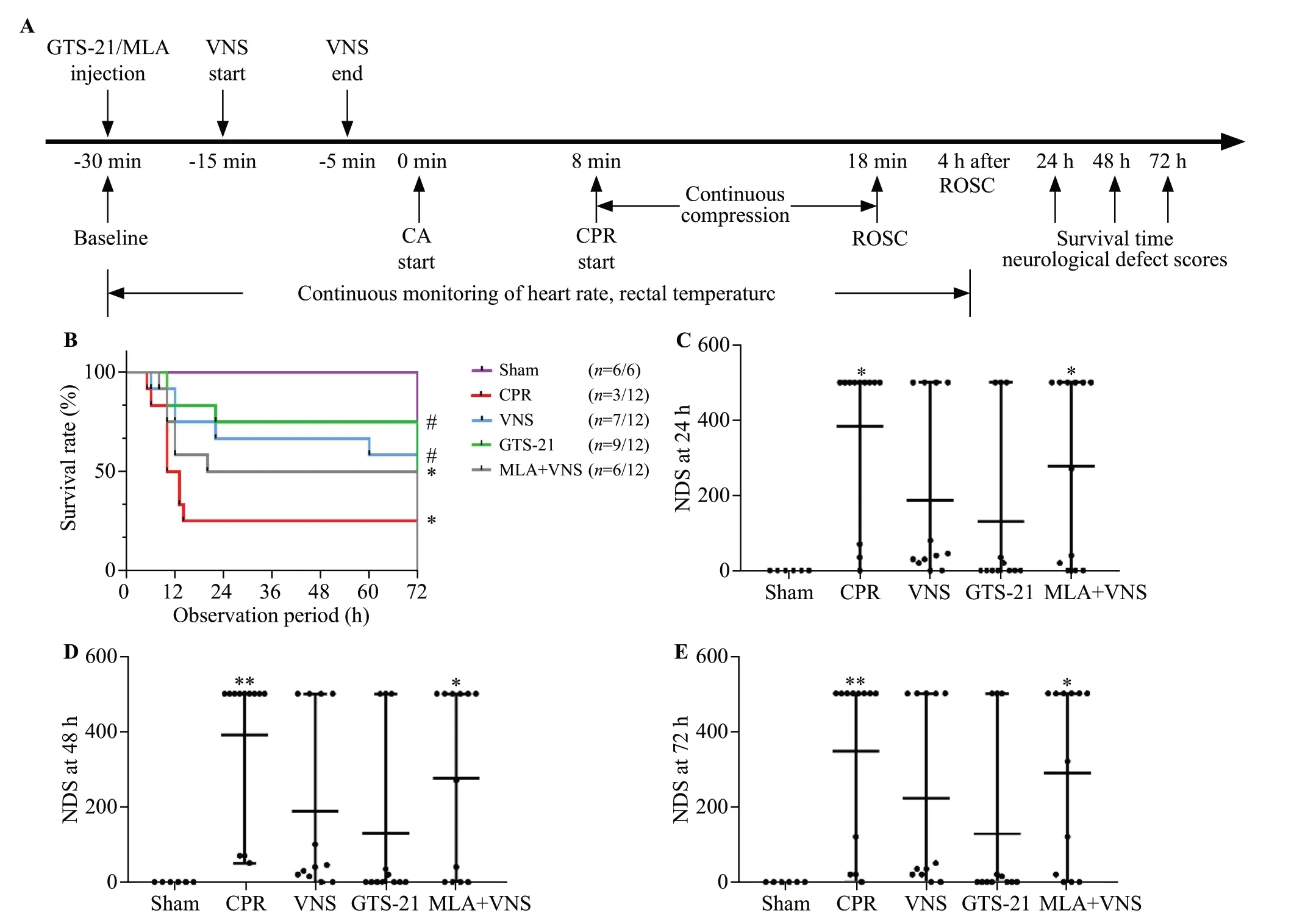
Figure 1.Vagus nerve stimulation (VNS) treatment improved 72-hour survival and neurological recovery in mice after cardiac arrest/cardiopulmonary resuscitation (CA/CPR).(A) experimental procedure.The GTS-21 (α7nAChR agonist) (4 mg/kg) was injected 30 min before CA.The methyllycaconitine citrate (MLA, inhibitor of α7nAChR) (4 mg/kg) was given 30 min before CA.(B) Kaplan-Meier curves of cumulative survival 72 h after CA and CPR in the five groups.VNS significantly improves neurological deficit score (NDS) at 24 h (C), 48 h (D), and 72 h (E)after CA/CPR.Black points indicate values for individual mice; horizontal bars indicate mean with range values (**P < 0.01 vs.Sham group; *P< 0.05 vs.Sham group; #P < 0.05 vs.CPR group).ROSC: return of spontaneous circulation.
DISCUSSION
In this study, the effect of VNS treatment after CA/CPR and its corresponding mechanism were investigated.The main findings are as follows: (1) VNS improved the 72 h survival rate and neurological recovery in mice after CA/CPR; (2) VNS alleviated the damage to hippocampal neurons and reduced the release of inflammatory mediators after CPR in mice; (3) nicotine attenuated the release of TNF-α in microglia after I/R and MLA reversed this protective effect; and (4) nicotine inhibited TLR4/NF-κB pathway by activating α7nAChR/JAK2 axis in BV-2 cells after I/R.
Studies on CAP show that α7nAChR activation plays a key role in protecting the brain.[22,23]In this study,we found that VNS improved the outcomes of CPR in a mouse model by activating α7nAChR-mediated CAP caused by intravenous potassium chloride.Unlike the mouse model in this study, the rat model of CA was produced by transcutaneous electrical epicardium stimulation in our previous study.[13]This experiment confirmed the protective effects of VNS in a small animal model with different CA mechanisms.Even so, its effect needs to be tested in a large animal model for further study.
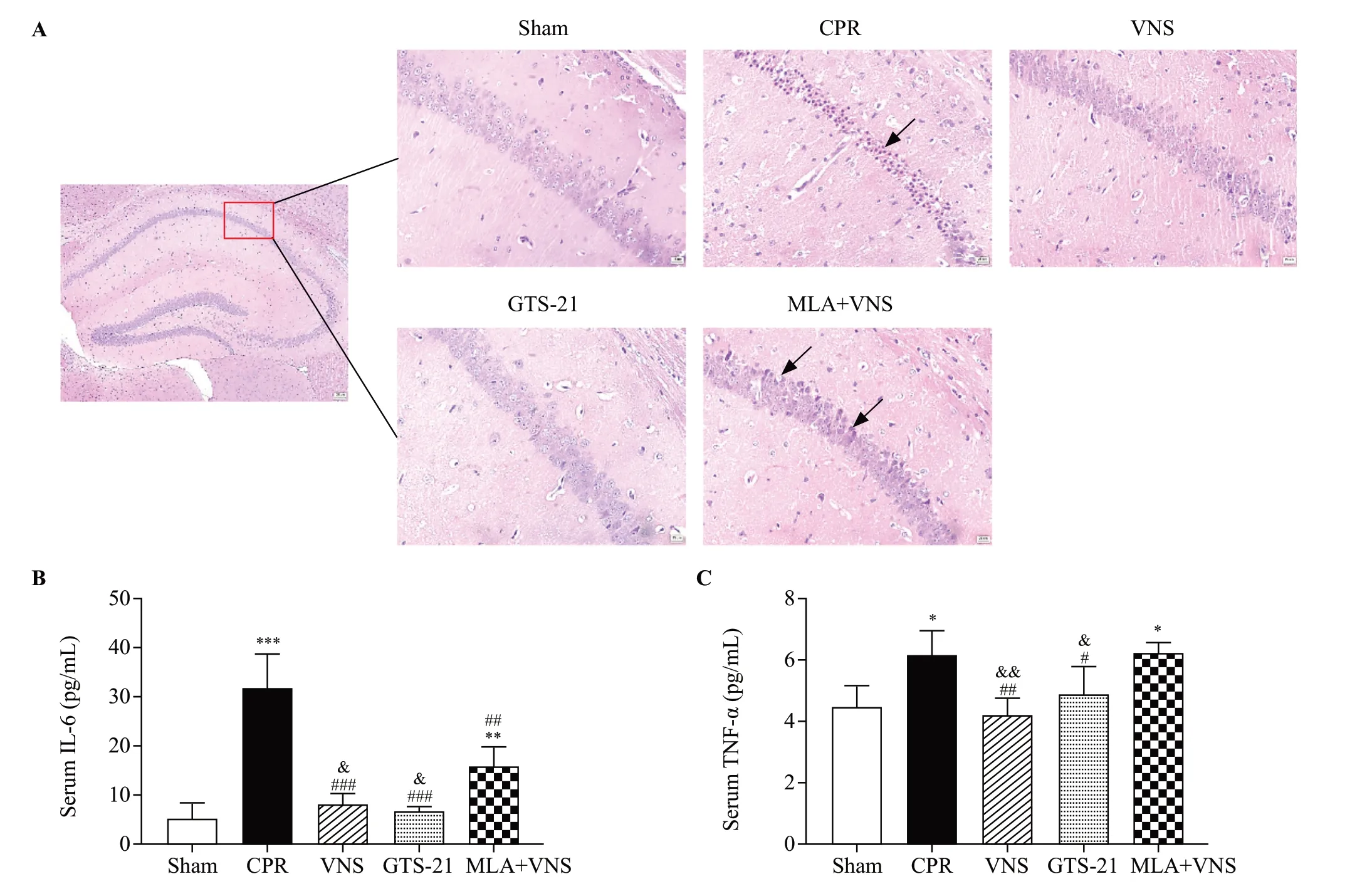
Figure 2.Vagus nerve stimulation (VNS) markedly reduced pathological damage in mice after cardiac arrest/cardiopulmonary resuscitation (CA/CPR).(A) HE staining in hippocampal CA1 region of mice at the 72 h after return of spontaneous circulation (ROSC).Red square shows the hippocampus CA1 region (magnification 40×).Right panels are 10× magnification photomicrographs from the red square of left panel.Black arrows indicate the nuclear pyknosis.Scale bar is 25 μm.The serum concentrations of interleukin-6 (IL-6) (B), and tumor necrosis factor-α (TNF-α)(C) at 72 h after resuscitation.(n=3, *P<0.05 vs.Sham group, **P<0.01 vs.Sham group, ***P<0.001 vs.Sham group; #P< 0.05 vs.CPR group,##P<0.01 vs.CPR group, ###P < 0.001 vs.CPR group; &P< 0.05 vs.MLA+VNS group, &&P< 0.01 vs.MLA+VNS group).

Figure 3.Nicotine attenuated the release of tumor necrosis factor-α(TNF-α) and methyllycaconitine citrate (MLA, inhibitor of α7nAChR)and AG490 reversed this effect.*P < 0.05.I/R: ischemia/reperfusion;Ni: nicotine.
To clarify the possible mechanism of VNS in this study, we measured the inflammatory cytokines in mice after resuscitation and found that VNS could reduce the expression of IL-6 and TNF-α induced by CA/CPR and this anti-inflammatory effect involved α7nAChR because MLA reversed this effect.Nonetheless, it was interesting to find that the anti-inflammatory effect via a7nAChR may be partly implicated in the association between VNS treatment and prolonged survival time because there was no significant difference in survival between the VNS group and MLA+VNS group.A possible explanation is that the role of VNS in improving the prognosis of CA/CPR is complicated, and involves factors besides inhibiting inflammation, such as anti-apoptosis and antioxidation effects.[21,24]
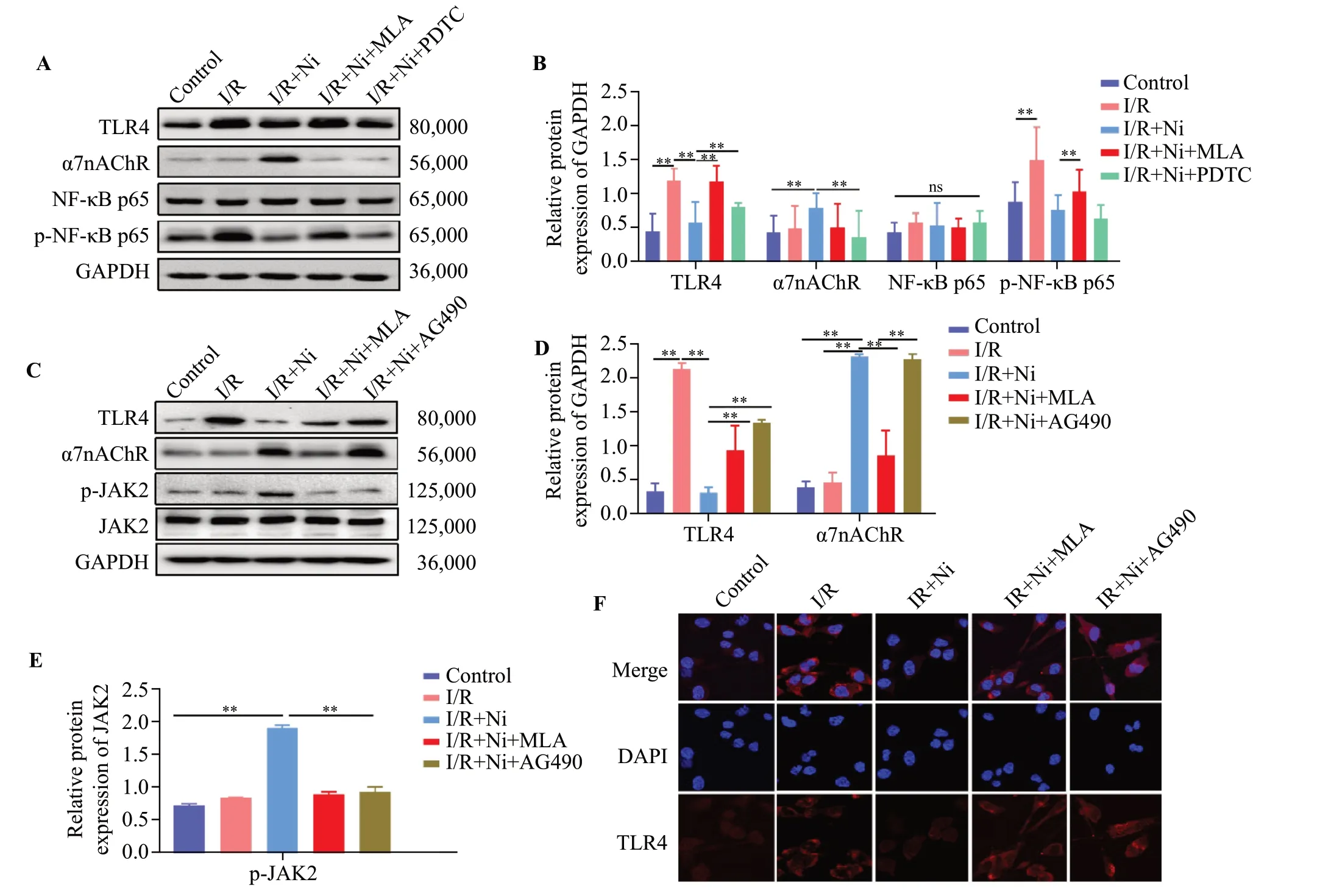
Figure 4.Nicotine inhibited TLR4/NF-κB pathway by activating α7nAChR/JAK2 axis in BV-2 cell after ischemia/reperfusion (I/R).(A)Representative immunoblots of TLR4, α7nAChR, NF-κB p65, and p-NF-κB p65.(B) Quantitative analysis of TLR4, α7nAChR, NF-κB p65,and p-NF-κB p65 expression.The protein amount was normalized to GAPDH.(C) Representative immunoblots of TLR4, p-JAK2, JAK2, and α7nAChR.Quantitative analysis of TLR4, α7nAChR (D) and p-JAK2 (E) expression.(F) Immunostaining of TLR4 in BV-2 cells.TLR4 (red),DAPI (blue).**P < 0.01; ns: not significantly different.
To further investigate the molecular mechanism of the protective effect of VNS, we used nicotine to activate the α7nAChR on BV-2 cell surface under OGD/R conditions,which mimic I/R injuryin vitro, and to study which downstream signaling molecules were involved.In a septic injury model, it has been found that stimulation of the CAP by nicotine improves the sepsis-induced mortality and this may be associated with inhibition of TLR4 expression via α7nAChR signaling.[15,25]In our I/R injury model,we also found that nicotine attenuated the increase in TLR4 protein expression, and this protective effect was abolished by MLA treatment, indicating that nicotine could protect against I/R injury by suppressing TLR4 expression through stimulation of α7nAChR.Although there is a crosstalk between TLR4 and α7nAChR after nicotine treatment, the molecular mechanism that regulates their expression is still unknown.JAK2 has been proven to be an early target of α7nAChR-mediated neuroprotection,[26]and activated α7nAChR can form a heterodimeric complex with JAK2, causing tyrosine phosphorylation, and thereby activating non-receptor tyrosine kinases.[27,28]Our results indicated that nicotine treatment increased the phosphorylation of JAK2 and the JAK2 inhibitor AG490 reversed the anti-inflammatory effect of nicotine, suggesting that the classical α7nAChR/JAK2 signaling pathway is involved in the present process.NF-κB is an important transcription factor that regulates the expression of proinflammatory cytokines in the signal transmission pathway.[29,30]Activation of α7nAChR may significantly block the NF-κB signaling pathway.[31,32]In addition, we found that nicotineactivated α7nAChR/JAK2 inhibited the expression of TLR4/NF-κB signaling pathway in BV-2 cells exposed to I/R, and pretreatment with MLA blocked this effect of nicotine.These results suggested that nicotine plays an anti-inflammatory effect by suppressing TLR4/ NFκB signaling pathway and activating α7nAChR/JAK2 signaling pathway in BV2 cells under OGD/R conditions.
However, there are some limitations to this study.First,this experiment adopted pre-treatment, which has a certain gap within clinical application.Second, we applied various blockers and agonists in our research, but the effect was not significant.The potential reason is that there are too many complicated pathways involved in the organism other than the specific pathways we want to observe.For this problem,it may be a better choice to adopt a corresponding gene knockout animal.In addition, other signaling pathways that are related to VNS such as α7nAChR/PI3k/Akt pathway were not investigated here.[33]
CONCLUSIONS
Our research shows that VNS can reduce inflammatory cytokines and attenuate brain injury in mice following CA, and its potential mechanism could be related to the activation of α7nAChR/JAK2 signaling pathway, and the inhibition of TLR4/NF-κB axis to alleviate the inflammatory response to I/R injury.Hence,VNS may be a therapeutic choice for the recovery of brain injury after CA.
Funding:This work was supported by research grants from the National Natural Science Foundation of China (grant no.81571866 and grant no.82072137).
Ethical approval:The present study was approved by the Medical Ethics Committee of Huazhong University of Science and Technology (Permit Number: S2356).
Conflicts of interest:The authors declare that they have no competing interests.
Author contribution:SX and LG contributed equally to this study, conceived of the study, conducted the experiments, collected the data, and drafted the preliminary manuscript.WJS and LCL participated and helped to perform the experiments.TTS and YHZ contributed to refining ideas and helped edit the manuscript.HH and GQG coordinated the data management, and participated in data acquisition and analysis.QZ and PS developed the study’s aims, interpreted the results and contributed to the completion of the manuscript.All authors read and approved the final version of the manuscript.All the supplementary files in this paper are available at http://wjem.com.cn.
杂志排行

World journal of emergency medicine的其它文章
- Tension urinothorax as a reversible cause of cardiac arrest: a case report
- A case of pulmonary mucormycosis presented with cardiac arrest
- Pyopneumothorax caused by Parvimonas micra and Prevotella oralis: a case report
- Hemorrhagic pancreatitis from fenofibrate and metformin toxicity: a case report
- The effect of prophylactic antibiotics in acute upper gastrointestinal bleeding patients in the emergency department
- The effects of hyperbaric oxygen therapy on paroxysmal sympathetic hyperactivity after cardiopulmonary resuscitation: a case series