微生物共培养促进植物乳杆菌RX-8 高效合成细菌素的调控机制
2023-11-26刘国荣刘洋硕聂蓉
刘国荣,刘洋硕,聂蓉
(1 北京工商大学食品与健康学院 北京100048 2 北京工商大学老年营养与健康教育部重点实验室 北京100048 3 北京工商大学北京市食品添加剂工程技术研究中心 北京 100048)
乳酸菌细菌素是在乳酸菌生长过程中自身产生的一种具有抗菌活性的多肽类物质,大部分乳酸菌细菌素具有高疏水性、高等电点以及阳离子性的特性[1-2]。与抗生素不同,细菌素可以被胰凝乳蛋白酶以及胃蛋白酶等人体中常见的蛋白酶降解,具有无毒、无抗药性、无残留等优点[3-4]。然而,细菌素的产量受多种因素的影响,例如培养条件、相关合成基因的表达、是否添加外源刺激物质等。由于外源物质的安全性较高且成本较低,因此目前的研究集中于构建外源微生物与产细菌素乳酸菌的共培养体系,以此提高细菌素的产量。有研究表明,外源微生物可以诱导乳酸菌细菌素的高效合成[5],Man等[6]研究发现嗜热链球菌STY-31 与植物乳杆菌KLDS1.0391 共培养会提高植物乳杆菌素(Plantaricin)MG 的产量;另外,Tabasco等[7]研究表明德氏乳杆菌保加利亚亚种CECT4005 和德氏乳杆菌乳酸亚种CECT282 等都会增强嗜酸乳杆菌La-5 产生乳杆菌素(Lactacin)B 的能力。
群体感应系统(Quorum sensing,QS)是一种细胞间相互交流的“语言”,分为种内和种间群体感应系统;信号分子随着细菌的生长不断产生,当积累到一定量后,便会被自身或其它细菌所感知,从而诱导特定基因的表达,使菌群表现出如形成生物被膜、产生细菌素等生理活动[8-9]。在纯培养中,植物乳杆菌素EF 的合成受到种内群体感应系统的调控,即:首先,植物乳杆菌自身通过plnA 基因编码合成信号分子自诱导肽(Autoinducing peptide,AIP);当细菌大量繁殖,种群密度不断增加致AIP 大量积累达到阈值时,激活位于细胞膜上的由plnB 基因编码的组氨酸蛋白激酶,并以磷酸化的方式将群体感应信号分子传递给相应的反应调节蛋白,这两个蛋白则分别由plnC 和plnD编码;接着调控操纵子plnEFI 产生植物乳杆菌素EF,并通过ABC 转运系统分泌到胞外[10-14]。而在共培养体系中,同时存在种间群体感应与种内群体感应两种信号分子。目前有研究发现植物乳杆菌DC400 和罗氏乳杆菌A7 在共培养过程中,LuxS基因的表达量上升,AI-2 的分泌量增加[15]。植物乳杆菌NMD-17 与瑞士乳杆菌NMD-137 进行共培养时,会显著增加AI-2 信号分子的活性,因此推测AI-2/LuxS 介导的种间群体感应系统在其中发挥一定的作用[16]。
本课题组发现植物乳杆菌RX-8 与枯草芽孢杆菌1.8715 共培养可以提高植物乳杆菌素EF 的产量。在阻断种间信号分子AI-2 后,会降低枯草芽孢杆菌1.8715 对共培养植物乳杆菌素EF 的诱导效应,说明种内群体感应系统参与调控共培养中植物乳杆菌素EF 的合成[17]。本研究基于以上结果,敲除种内群体感应系统关键基因信号分子AIP 的编码基因plnA,将突变菌株植物乳杆菌ΔRX-8、野生菌株植物乳杆菌RX-8 分别与枯草芽孢杆菌1.8715 共培养后,测定信号分子的分泌情况及相关基因的表达量,探究群体感应系统调控细菌素合成的作用机制。这对于在共培养体系中提高乳酸菌细菌素的产量,以及群体感应相关基因的研究提供理论依据,为乳酸菌细菌素的工业化生产提供技术支持。
1 试验材料
1.1 菌株与质粒
本研究所用的菌株和质粒见表1。

表1 菌株和质粒Table 1 Bacterial strains and plasmids
1.2 试剂
Q5 高保真DNA 聚合酶,购于北京欣华绿源科技有限公司;E.Z.N.A 质粒小提试剂盒,购于上海索宝莱科技有限公司;ClonExpress II 克隆试剂盒,购于南京诺唯赞生物技术有限公司;红霉素、氨苄青霉素、DNA marker、多功能DNA 纯化回收试剂盒,购于北京半夏生物科技有限公司;限制性内切酶,购于Takara 公司。
1.3 仪器与设备
C1000 Touch PCR 仪,美国BIO-RAD2 公司;SPX-150B 生化培养箱,上海博讯医疗生物仪器股份有限公司;BSA224S 型数字电子天平,德国Sartorius 公司;涡旋振荡器,德国IKA 公司;小型高速离心机,美国SIGMA 公司;BioSpectrum 凝胶成像仪,美国UVP 公司;UB-7 pH 计,美国DENVER 仪器公司;电穿孔仪,德国Eppendorf 公司。
1.4 引物设计
利用Primer Premier 5.0 进行引物设计,如表2 所示。

表2 引物列表Table 2 Primer design
2 试验方法
2.1 基因缺失菌株及种内群体感应系统缺失共培养体系的构建
2.1.1 质粒pMG36e-red/cas 的构建 从含有pCas 的大肠杆菌(Escherichia coli)DH5α 中提取质粒,操作步骤参考试剂盒说明书,以质粒pCas为模板,使用引物Cas9-F/R 和Red-F/R 扩增CRISPR/Cas9 基因编辑所需的敲除元件内切酶基因Cas9 和重组修复酶基因λ-Red。
利用TaKaRa Quickcut SacI、KpnI 限制性内切酶切质粒pMG36e,将环形pMG36e 质粒线化,所得酶切产物电泳验证并回收。将切胶回收后的线性质粒pMG36e 和纯化后的Cas9 和λ-Red 片段进行多片段一次性连接。将得到的产物与感受态细胞混匀冰上静置,随后热激冰浴并加入LB 培养基。37 ℃充分复苏后,取菌液涂布于氨苄青霉素抗性LB 平板上,37 ℃培养24 h。
2.1.2 sgRNA 的设计 根据植物乳杆菌RX-8 种内信号分子编码基因plnA 的序列和靶位点设计原则,选择带有PAM(NGG)位点的位置设计靶向sgRNA,命名为plnA-sgRNA,并以质粒pNZ8148来源的PNis 启动子作为sgRNA 的启动子,诱导表达sgRNA。
2.1.3 plnA 同源臂片段的扩增及与质粒的连接转化 分别以plnA-L F/R 和plnA-R F/R 为引物,以植物乳杆菌RX-8 基因组为模板扩增plnA 基因两侧同源臂。将纯化回收的同源臂片段和线化的质粒连接并转化入大肠杆菌DH5α。
2.1.4 敲除质粒pMG36e-red/cas-sgRNA/plnALR 构建 将线化质粒pMG36e-red/cas、pNissgRNA 和同源臂片段plnA-LR 进行多片段一次性连接,连接后转化入大肠杆菌DH5α 进行酶切并测序验证。
2.1.5 敲除菌株乳杆菌RX-8ΔplnA 构建 将原始菌株RX-8 活化两代后,接种至含2%甘氨酸的MRS 培养基中,静置培养至对数生长期,加入氨苄青霉素(终质量浓度为10 μg/mL),继续培养至其OD600nm在0.4~0.5。离心收集菌体细胞。用预冷的电穿孔缓冲液清洗细胞2 次后,加入预冷的500 μL 30% PEG-1500 重悬菌体,得到感受态细胞。
将上述得到的质粒DNA 加入感受态细胞中,移液枪吹吸混匀后冰浴5 min,转移至提前预冷的电转杯,设置电击参数为:电压2.5 kV,电脉冲25 μF,电阻200 Ω。电击后立即用含2 mmol/L CaCl2和20 mmol/L MgCl2的预冷的MRS 肉汤稀释,并在30 ℃下孵育3 h。吸取200 μL 到红霉素MRS固体平板上,涂布后培养24 h。挑取生长情况良好的单个菌落,分别接种于具有氨苄青霉素抗性和自诱导肽的LB 试管中,37 ℃,200 r/min 摇菌扩增12 h 后,提取植物乳杆菌RX-8 基因组DNA。用引物ΔplnA-F/R 扩增敲除片段,将PCR 产物测序与野生菌株序列进行对比。
2.2 共培养中突变菌株的产细菌素变化分析
2.2.1 突变菌株生长曲线的测定 将野生菌株RX-8 和敲除菌株ΔRX-8 接种到MRS 培养基中,并将枯草芽孢杆菌1.8715 的种子液等比例分别接入,形成共培养体系后连续培养32 h,每隔4 h取样测OD600nm,绘制生长曲线。
2.2.2 突变菌株抑菌活性的测定 采用硫酸铵沉淀法制备[18]细菌素粗提液,采用琼脂扩散法[19]进行测定抗菌活性。在培养的固体平板上观察是否具有明显的抑菌圈,并用游标卡尺测量抑菌圈的直径抑菌圈直径越大,抑菌效果越好。
2.3 共培养体系中种间信号分子AI-2 的分泌量检测
从野生菌株RX-8 和敲除菌株ΔRX-8 种子培养液中取菌液按照1%接种量接种到MRS 培养基中37 ℃培养32 h,离心收集上清液。从BB170种子培养液中取菌液按照1%接种量接种到AB培养基,30 ℃培养至OD600nm在0.7~1.2,稀释后得到BB170 稀释液,并以BB170 荧光值为对照,计算野生菌株和突变菌株的相对发光值。
2.4 共培养中突变菌株相关基因的转录水平研究
2.4.1 突变菌株与野生菌株总RNA 提取及反转录cDNA 的制备 将野生菌株RX-8、突变菌株ΔRX-8 进行纯培养并分别与枯草芽孢杆菌1.8715 共培养提取每隔4 h 的野生菌株与突变菌株的发酵液,离心收集菌体,采用RNA 提取试剂盒得到总RNA,并利用cDNA 合成试剂盒提取制备cDNA。
2.4.2 RT-qPCR 分析 对共培养中野生菌株RX-8 和突变菌株ΔRX-8 的ABC 系统转运基因LuxS、甲硫腺苷核苷酶基因pfs、群体感应基因plnBCD、细菌素基因plnEF 的表达量进行测定。测定结果以纯培养时的RX-8 基因表达量为基准(即为1)进行相对表达量的计算。
3 结果与分析
3.1 构建种内群体感应缺失的共培养体系
3.1.1 质粒pMG36e-red/cas 的构建 本研究以质粒pMG36e 为敲除骨架,插入CRISPR/Cas9 基因编辑所需的组件,初步构建了敲除工具。如图1所示,在第1 泳道和第2 泳道分别扩增得到2 个位于3~5 kp 和2~3 kp 之间的DNA 片段,与预计扩增片段大小(4 107,2 210 bp)基本一致,并且测序结果显示序列完全正确。将线化质粒pMG36e与敲除组件Cas 和λ-Red 连接转化,对重组质粒进行酶切(XhoI、BglII 和BamHI、XhoI)验证,第3泳道为重组质粒pMG36e-red/cas(大小约为8.8 kb),第4 泳道的cas9 片段和pMG36e-λ-Red 分别实际长度为5 820 bp 和4 107 bp,测序结果比对正确,表明成功构建质粒pMG36e-red/cas。

图1 质粒pMG36e-red/cas 的构建电泳图Fig.1 Construction of plasmid pMG36e-red/cas
3.1.2 sgRNA 的设计 根据https://links.jianshu.com 网站在线设计plnA 基因的sgRNA。在网站中导入plnA 基因后可生成所有可能的靶点位置,选取得分最高的序列。本研究选择距离启动子ATG只有17 碱基的位置并且尾端带有PAM 序列的sgRNA 序列,以pNZ8148 来源的pNis 启动子作为sgRNA 的启动子,诱导其表达。目的基因plnA 中sgRNA 所在位置及其碱基序列如图2 所示。将设计好的plnA 基因的sgRNA 序列和pNis 启动子序列构成的pNis-sgRNA 进行合成,电泳结果如图3所示。

图2 plnA 基因的sgRNA 位置及序列Fig.2 Insertion position of sgRNA and sequences of plnA
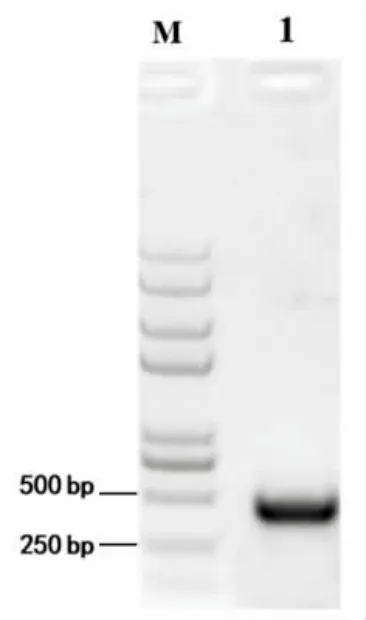
图3 pNis-sgRNA 酶切电泳图Fig.3 Agarose gel electrophoresis pattern of RCR amplification product of pNis-sgRNA
3.1.3 plnA 同源臂的设计 扩增plnA 基因左右两端的同源臂plnA-L(493bp)和plnA-R(526 bp),图4 中显示分别扩增得到2 个位于500~750 bp 之间的DNA 片段,与预计扩增片段大小(500 bp)条带大小相符且测序结果显示序列完全正确,表明扩增成功。接着将纯化回收后的同源臂和质粒分别连接,酶切鉴定结果图如4 所示。重组质粒经过XhoI 和SpeI 双酶切后在1~2 kb 之间可见1 000 bp 左右的条带,电泳结果与测序结果均表明与预期相符。

图4 plnA 同源臂L/R 基因的PCR 产物和双酶切电泳图Fig.4 A garose electrophoresis of PCR products of plnA-L/R
3.1.4 敲除质粒pMG36e-red/cas-sgRNA/plnALR 的构建 将质粒pMG36e-red/Cas 与同源臂plnA-L/R、pNis-gRNA 连接转化。如图5 所示,经XhoI 和SpeI 双酶切后可见在1~2 kb 和8 kb 出现两条特异性条带,均与预期大小一致。将重组的敲除质粒和酶切片段测序,结果与原序列一致,分别为同源臂片段1 019 bp 和pMG36e-red/cas-sgRNA 10 680 bp。
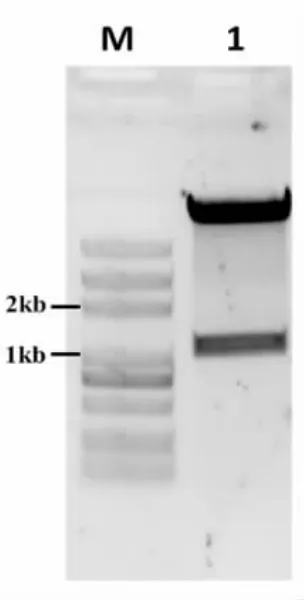
图5 敲除质粒酶切PCR 产物电泳图Fig.5 Electrophoretic pattern of knockout plasmid enzyme digestion PCR products
3.1.5 突变菌株ΔRX-8 的构建 将敲除质粒CRISPR/Cas-sgRNA-plnA-L/R 电击转化入植物乳杆菌RX-8 中,通过引物ΔplnA-F/R 扩增靶基因的同源臂,判定plnA 基因的敲除情况。由于敲除片段长度较短在电泳图中不明显,这里通过测序序列峰图和序列对比图展示敲除结果如图6,从峰图中可见敲除plnA 基因后进行了同源修复并引入设计的强终止子序列。
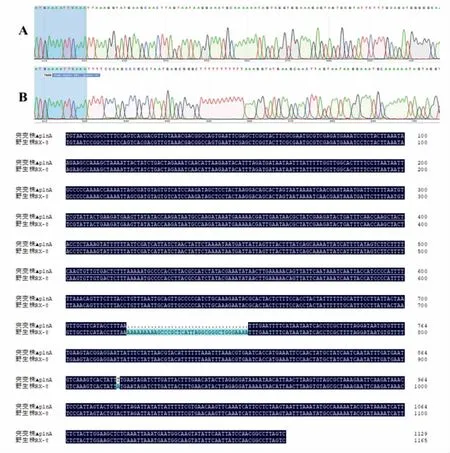
图6 plnA 基因敲除前后的测序鉴定结果Fig.6 Sequencing results before and after plnA gene knockout
3.2 共培养中突变菌株的产细菌素变化
3.2.1 共培养中突变菌株生长情况 为研究在共培养中种内群体感应系统关键基因信号分子plnA基因敲除后植物乳杆菌RX-8 的生长代谢变化,本研究绘制了共培养32 h 的念珠菌的生长曲线图。如图7 所示,在敲除了plnA 基因之后,无论是纯培养还是共培养过程中,突变菌株仍然保持着良好的生长活性,且共培养的菌体密度高于纯培养。与野生菌株相比,突变菌株的菌体密度整体略低,但是并无明显的差异,依旧保持平稳的生长趋势。

图7 野生菌株RX-8 和突变菌株ΔRX-8 的生长曲线Fig.7 Growth curve of wild plant RX-8 and mutation plant ΔRX-8
3.2.2 共培养中突变菌株细菌素合成情况 野生菌株RX-8 和和突变菌株ΔRX-8 在纯培养和共培养体系中细菌素合成量如图8 所示。纯培养体系中,由于敲除了ΔplnA,突变菌株不会产生细菌素,所以抑菌圈直径为0。在共培养体系中,突变菌株植物乳杆菌ΔRX-8 的细菌素合成量虽较野生菌株植物乳杆菌RX-8 有所下降,但却明显高于纯培养体系中野生菌株植物乳杆菌RX-8 细菌素的合成量,说明敲除plnA 后依然部分存在枯草芽孢杆菌1.8715 的诱导作用。从整体来看,细菌素在整个培养时期均表现出先上升后下降的趋势,且纯培养体系中细菌素的产量以及合成速度均高于纯培养体系。

图8 细菌素合成量变化Fig.8 The changes in bacteriocin synthesis
3.3 共培养中突变菌株群体感应系统信号分子分泌情况
敲除种内信号分子基因plnA 后,研究共培养过程中野生菌株RX-8 与突变菌株ΔRX-8 种间信号分子AI-2 分泌量,结果如图9 所示。以上清液的相对发光量作为AI-2 分泌量的判断依据,在4~20 h 的培养过程中,纯培养体系AI-2 分泌量显著低于共培养体系,但是在20 h 之后便无明显差异。从菌株水平来看,在整个培养过程中,无论是共培养还是纯培养,突变菌株与野生菌株AI-2的分泌量始终保持在同一水平。
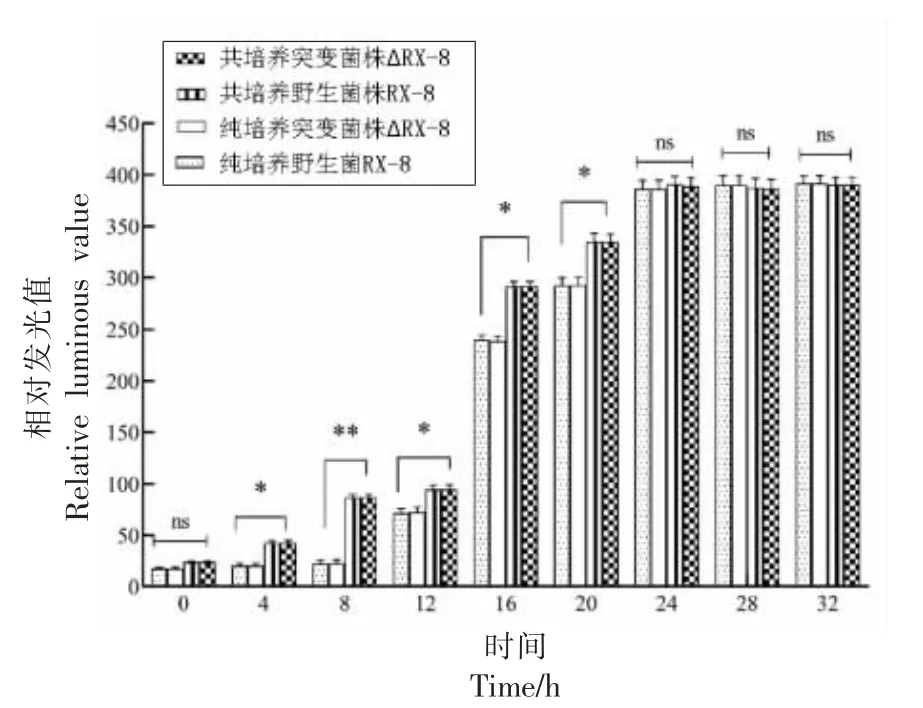
图9 种间信号分子AI-2 分泌情况Fig.9 Secretion of interspecies signaling molecule AI-2
3.4 共培养体系中突变菌株群体感应系统相关基因的表达情况
3.4.1 对种间信号分子合成基因LuxS、pfs 的影响 对种间信号分子合成相关基因LuxS、pfs 的相对表达量进行测定,结果如图10 所示。在整个培养的过程中,共培养体系的突变菌株与野生菌株在LuxS、pfs 的表达量上均未有明显差异,但是与纯培养相比表达量均有所上升。由此可以看出敲除种内信号分子编码基因plnA 对种间信号分子合成基因的转录无影响,这与AI-2 分泌量未发生变化的结果一致。
3.4.2 共培养中突变菌株群体感应系统双组分系统基因表达情况 敲除种内信号分子基因plnA后,研究共培养过程中野生菌株RX-8 与突变菌株ΔRX-8 的群体感应系统中双组分系统编码基因plnBCD 的转录水平,结果如图11 所示。相对表达量是以纯培养过程中植物乳杆菌RX-8 的基因表达量为基准,因此相对表达量大于1 即为基因表达上调。由图11 可知,在敲除了plnA 之后,基因plnBCD 的相对表达量均大于1,这意味着相关基因仍然表达且处于上调水平,由此推测在共培养的过程中可能存在某些物质刺激相关的受体蛋白,从而调控相应基因的表达。与野生菌株相比,突变菌株的组氨酸激酶编码基因plnB 表达量有所下降,差异主要集中在菌株生长的对数期及稳定期,即12~32 h;而作为下一级信号基因的plnCD,表达差异略有滞后且时间缩短,集中在16~28 h。从整体上看,3 个基因的相对表达量变化与细菌素的产量具有相同的变化趋势,呈现出先上升后下降。

图11 双组分系统基因plnBCD 表达情况Fig.11 Changes in expression of two-component system gene plnBCD

图12 细菌素结构基因plnEF 表达量变化Fig.12 Changes in expression of bacteriocin structural gene plnEF
3.4.3 对细菌素合成基因表达的影响 从基因的转录水平上可以看出,细菌素合成基因plnEF 的表达量有显著差异,在敲除了plnA 基因之后,突变菌株的plnEF 表达量明显低于野生菌株,但是相对表达量仍然大于1,与纯培养相比基因表达上调。共培养体系中,0~4 h 由于在细菌处于迟滞期,细菌素并未开始合成,因此两菌株的基因表达量并无显著差异;从8 h 开始进入对数生长期,菌体密度开始增加,同时细菌素开始合成,相关的细菌素结构基因开始表达,导致二者的表达量开始具有显著差异直至培养结束。
4 讨论与结论
本研究对采用CRISPR/Cas9 基因敲除技术构建突变菌株植物乳杆菌ΔRX-8,将其与枯草芽孢杆菌1.8715 进行共培养,同时以野生菌株RX-8与枯草芽孢杆菌1.8715 的共培养体系为对照,研究种内群体感应系统在枯草芽孢杆菌1.8715诱导植物乳杆菌RX-8 细菌素产生增加过程中所起的作用以及相关基因的变化情况。
根据敲除菌株与野生菌株的生长曲线结果显示,在敲除了plnA 基因之后,植物乳杆菌RX-8仍然可以正常生长。共培养体系和纯培养体系中,两者的菌体密度并无显著差异,具有相同的生长趋势,这表明plnA 基因是一种非致死性基因,敲除之后对植物乳杆菌RX-8 生长情况没有明显的影响,可以排除因为生长情况而导致细菌素产量下降的干扰因素,同时也说明种间信号分子缺失共培养体系的成功构建。
从细菌素的合成情况可以发现,相较于纯培养体系中的野生菌株RX-8,共培养体系中突变菌株ΔRX-8 的细菌素产量显著上升,但是仍然低于野生菌株的共培养体系,这说明细菌素的诱导合成途径并不完全是由种内群体感应系统所调控的,种间群体感应系统在细菌素的合成过程中同样发挥作用。而种间信号分子AI-2 分泌量的结果显示,当细菌处于对数生长期时,共培养体系与纯培养体系的分泌量具有显著差异,待进入稳定期后便无明显区别。在共培养过程中,敲除菌株与野生菌株种间信号分子AI-2 的分泌量始终保持一致,且在纯培养体系中亦是如此。上述结果均表明在种内群体感应系统的缺失的共培养体系中,种间群体感应系统信号分子的分泌不会受到影响。
从转录水平上看,在共培养体系中,敲除了plnA 基因之后,调控双组分系统plnBCD 基因的表达量略低于野生菌株,但是依旧属于正常水平。共培养体系中组氨酸蛋白激酶编码基因plnB 的表达量略有降低,这是因为构建种内群体感应系统缺失共培养体系后,突变菌株ΔRX-8 不再分泌种内信号分子AIP,而组氨酸蛋白激酶恰好是AIP的受体蛋白,导致受体蛋白只能被部分激活[20-22]。在Zhang等[23-24]的研究中,敲除了plnB 基因之后的突变菌株植物乳杆菌ΔLXM-1 的细菌素产量明显降低,在补充PlnA 之后仍然无法恢复,这是由于在缺乏受体蛋白的情况下,即使有信号分子PlnA 也无法激活种内群体感应系统来调控细菌素合成。而在本研究中,缺失了PlnA 之后,plnB 的表达量虽然有所下降,却不会降低至纯培养水平,说明了PlnB 可能被其它信号分子激活。另外,与纯培养的野生菌株相比,共培养的野生菌株和突变菌株种间信号分子AI-2 的分泌量明显增加,同时基因LuxS,pfs 的表达量均有上调。姜雪雍等[25]的研究结果显示,在副干酪乳杆菌HD1.7 与枯草芽孢杆菌构成的共培养体系中,细菌素的合成量是纯培养组的1.21 倍,群体感应相关基因LuxS上调2.31 倍。而在孙思睿等[26-28]的研究中,纯培养体系下组氨酸蛋白激酶基因细菌素结构基因plNC8HK、双组分调节基因plnD 以及细菌素结构基因plnEF 均因LuxS 基因的缺失,表达显著下调,这与本研究结果一致。因此推测种间信号分子AI-2 可能与AIP 共用同一套双组分系统。在共培养12 h 时,二者plnB 基因的表达量已经具有显著差异,但是其下级传导基因的表达量虽有差异却并不显著,直至16 h 时差异开始显著,因此存在基因表达的滞后性[29-31]。同时,细菌素合成基因plnEF 的表达量有明显差异,突变菌株ΔRX-8 的plnEF 基因的表达量比野生菌株略有降低,但细菌素的产量仍然高于纯培养中的野生菌株,这与细菌素表达量的变化趋势相符。
综合以上结果,在种内信号分子缺失的共培养体系中,诱导效应并未因为缺失PlnA 而完全丧失,表明除PlnA 介导的种内群体感应系统之外,还有其它系统调控诱导效应。由于在种内群体感应系统缺失的共培养体系中不会影响种间信号分子AI-2 的合成,而结果显示AI-2 的分泌量提高,推测可能是存在种间信号分子AI-2 与种间信号分子AIP 共用同一套双组分系统的现象,两种信号分子共同促进细菌素的高效合成。
