极光激酶B 抑制剂的研究进展
2023-11-24马澜婧杜海琛张百红
马澜婧,杜海琛,张百红
中国人民解放军联勤保障部队第940 医院 肿瘤科,甘肃 兰州 730050
极光激酶(AURK)是维持细胞基因组完整性的关键丝氨酸/苏氨酸有丝分裂调节激酶,由3 个成员极光激酶A、B、C 组成[1]。其中极光激酶B(AURKB)由位于17 号染色体上的AURKB 基因编码,是染色体载体复合体(CPC)的激酶模块组成蛋白,在有丝分裂中起着至关重要的作用[2]。该复合体定位模块还包括内着丝粒蛋白(INCENP)、北极素和生存素[3]。AURKB 在有丝分裂过程中最活跃。有丝分裂启动前,AURKB 广泛分布在染色体臂上,通过组蛋白H3 和中心体蛋白A 的磷酸化促进染色体浓缩[4-5]。在有丝分裂前中期阶段,AURKB 作为CPC 的一部分,移动至着丝粒处,并保持在这个位置,一旦细胞分裂,AURKB 将进一步迁移到中心纺锤体[6-7]。AURKB 已被证明可以调节着丝粒激活纺锤体组装检查点[8-9]。并且AURKB受小泛素样修饰物、去泛素化酶、类端粒沉默干扰体1 和赖氨酸特异性去甲基酶等多种表观遗传修饰酶调节[10-13]。AURKB 确保染色体的充分对齐和分离,并在中期到后期过渡期间重新定位到微管[14]。研究表明,细胞对着丝粒处的低张力敏感,并通过主动募集AURKB 进行纠错来做出反应[15]。最新的研究提示,AURKB 也可通过调节复制蛋白A 维持基因组稳定性[16]。而调节复制蛋白A 亦可以通过人类肿瘤抑制因子环指蛋白20 介导的组蛋白H2B 单泛素化途径确保着丝粒处AURKB 的适当激活和DNA 断裂处修复蛋白的有效负载[17]。
上述生理过程也受外源性因素影响。研究发现城市灰尘颗粒可通过失活有丝分裂早期着丝粒处的AURKB 功能破坏有丝分裂进程[18]。孕酮受体膜成分1 可通过调节AURKB 和纺锤体、CPC 之间的联系影响卵泡生长[19]。HIV-1 包膜蛋白和CD4 相关作用诱导AURKB 重新定位到着丝粒,该作用与HIV 在细胞间融合和传播活性有关[20]。致癌基因v-Src可通过间接抑制AURKB 活性而使AURKB 离域[21]。丙型肝炎病毒感染人肝癌Huh-7.5 细胞可导致AURKB 活性降低,影响炎症途径[22]。而4-苯氧基喹啉衍生物可通过破坏AURKB 的有丝分裂定位实现抗肿瘤作用[23]。淋巴细胞抗原6K 通过AURKB及其底物组蛋白H3 信号轴发挥促癌作用[24]。
在非有丝分裂的情况下,AURKB 也被证明可以调节端粒酶来维持端粒,非有丝分裂相关地调节组蛋白H3 的状态,以及调节染色质重塑[25-27]。有研究表明AURKB 抑制剂对葡萄膜黑色瘤的抗肿瘤作用即与该功能有关[28]。
AURKB 在多种恶性肿瘤中过表达,并且在所有过表达AURKB 的病变中,组蛋白H3 的磷酸化都可以清楚地检测到,并且AURKB 失调会产生严重的细胞内后果,故AURKB 被作为有吸引力的抗癌药物靶点被广泛研究。极光激酶均包含3 个不同的结构域:可变N-末端结构域(39~139 个氨基酸)、保守激酶催化结构域(250~300 个氨基酸)和短C-末端结构域。立体结构上AURKB 与INCENP 形成复合体,见图1。AURKB 具有经典的双叶蛋白激酶折叠结构,其中富含β 链的N-末端结合域与核苷酸结合有关,并与激酶调节因子相互作用;C-末端结构域主要是α-螺旋的,用作底物的对接位点,并含有直接磷酸转移的残基;而ATP 结合口袋位于瓣叶之间的界面处[29],其内核苷酸的存在有利于晶体接触[30]。故AURKB 的ATP 结合口袋内是小分子抑制剂的理想靶点。主要原理为抑制性底物模拟效应,即抑制蛋白以ATP 相关的方式通过与蛋白底物竞争以高亲和力结合到蛋白激酶的催化亚基。具体来说ATP 竞争性AURKB 抑制剂结合到AURKB 的C末端结构域,从而抑制AURKB 催化活性[30]。抑制AURKB 的激酶活性可以阻止染色体排列和分离,进而阻止细胞分裂,也可以直接抑制胞质分裂;此外通过超越纺锤体检查点,这些细胞在正常时间内退出有丝分裂,迅速变成四倍体;并且由于AURKB不会阻断细胞周期的进展,这些高度异常的细胞在存在大量基因组不稳定的情况下继续增殖,迅速导致细胞死亡[31],这是AURKB 抑制剂最核心的抗癌原理。AURKB 主要在有丝分裂过程中表达和激活,非增殖细胞不会受到这些药物的不利影响。考虑到体内大多数正常细胞不会快速增殖,AURKB 抑制剂可能比非特异性细胞毒性药物具有更好的应用前景。
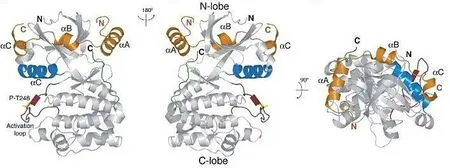
图1 AURKB:INCENP 立体结构Fig.1 Stereo-structure of AURKB:INCENP Complex
目前已经开发了多种靶向AURKB 的小分子抑制剂,均可以抑制AURKB 的自身磷酸化和组蛋白H3 的磷酸化。由于极光激酶家族的成员在激酶结构域中具有高度同源性,AURK 抑制剂的活性大多重叠,针对AURKB 的特异性抑制剂较少,有待进一步开发。本文介绍了AURKB 特异性抑制剂、进入临床试验阶段的泛AURK 抑制剂等的研究进展,希望为AURKB 抑制剂的开发和临床使用提供参考。
1 AURKB 特异性抑制剂
1.1 巴拉塞替
巴拉塞替是通过优化ZM447439 开发的基于喹唑啉衍生物的ATP 竞争性AURKB 抑制剂,包括AZD1152、AZD1152-HQPA 和AZD2811 3 种亚型,半数最大抑制浓度(IC50)为0.37 nmol/L(无细胞测定法)或1 nmol/L(激酶测定法)[32],与AURKA相比,它对AURKB 的亲和力高出1 000 倍以上[33],是第一个进入临床试验并已开展试验项目最多的AURKB 选择性抑制剂。巴拉塞替已证实对多种肿瘤有抑制增殖和/或诱导凋亡的作用。比较有代表性的有巴拉塞替对T790M 阴性非小细胞肺癌细胞系有强大的抗增殖作用,IC50<0.06 µmol/L[34]。体内外实验均证实巴拉塞替可抑制小细胞肺癌肿瘤生长,IC50<50 nmol/L,且生长抑制程度与癌基因CMYC表达水平呈正相关[35]。进一步研究发现AURKB 通过在Ser67 处磷酸化来稳定C-MYC,然后C-MYC 激活AURKB 转录,形成正反馈回路,这是AURKB 重要的促癌机制[36]。最新的研究发现,AZD1152 对胶质母细胞瘤原代培养细胞有杀伤作用,IC50为25 nmol/L,并且联合肿瘤电场治疗具有协同增效作用[37]。体内实验证实,300 nmol/L AZD1152 足以抑制人宫颈癌细胞系(C33A、HeLa和Caski)的分化和存活;裸鼠成瘤试验中ig 给药50 mg/kg AZD1152(隔日1 次,连续6 次)可显著减少HeLa 细胞肿瘤体积[38]。
1 项Ⅱ期临床试验表明,急性髓系白血病第1~7 天静滴AZD1152 1 200 mg,每28 天重复,治疗的总体缓解率为46%[39]。1 项Ⅱ期临床试验中,96 h 持续静滴AZD1152 800 mg,每21 天重复,对于B 细胞淋巴瘤仅有相对较低的总体缓解率(20%),提示AZD1152 不适合单药治疗[40]。在泛进展期实体瘤的研究中,AZD1152 最大耐受剂量为150 mg、48 h 持续静滴或第1~2 天静滴110 mg,均为每14天重复;尽管23%的患者治疗后疗效评价为稳定,但没有完全缓解或部分缓解的病例,总体AZD1152单药治疗疗效欠佳[41]。以上研究证实AZD1152 不良反应可控,中性粒细胞减少症是最常见的,且是剂量限制性毒性。AZD1152 的给药模式是多日静脉给药或静脉连续输注,缺乏便利性。因此促进了AZD1152 纳米颗粒制剂的开发,即AZD2811。
研究显示AZD2811 不仅不良反应更小,给药模式更便利,且抗肿瘤活性超过了AZD1152[42-43]。2017 年阿斯利康启动了AZD2811 作为单一疗法或联合疗法治疗无法耐受强化治疗的幼稚或复发/难治性急性髓性白血病的研究,旨在测试AZD2811 的最大耐受剂量[44]。可惜由于阿斯利康公司的战略调整,该试验于2021 年提前终止。另1 项AZD2811用于晚期实体瘤患者的安全性、耐受性和药动学的1 期研究表明,AZD2811 最常见不良事件为小剂量(≤200 mg/周期)时为疲劳(27.3%),大剂量时(≥400 mg/周期)为中性粒细胞减少症(37.9%),中性粒细胞减少症亦为剂量限制性毒性,最大耐受剂量为500 mg(第1 天静滴、每21 天重复),总体耐受性良好,疗效评定部分缓解率和稳定率分别为2.0%、45.1%[45]。
体外实验确认AZD2811 对小细胞肺癌细胞株普遍抗肿瘤活性良好后,3 项AZD2811 针对小细胞肺癌的临床试验应运而生[46]。1 项AZD2811 单药作为小细胞肺癌二线或三线治疗的临床试验,疗效评定33.3%的病例为稳定,提前达到研究目的终止[47]。另有2 项AZD2811 联合度伐利尤单抗治疗小细胞肺癌的试验,结果尚未发布。最新的研究在57 种小细胞肺癌细胞系和人源异种移植物模型中,验证AZD2811 的生长抑制活性,发现对AZD2811 敏感的亚群通常以但不限于高C-MYC基因表达为特征,重要的是B 细胞淋巴瘤2(BCL2)基因的高表达能够预测小细胞肺癌对AURKB 抑制剂反应的耐药性,与C-MYC基因状态无关;AZD2811(30 nmol/L)诱导的DNA 损伤和细胞凋亡受到高BCL2 水平的抑制,AZD2811(100 nmol/L)与BCL2 抑制剂维奈妥拉联合使用可显著致敏耐药模型;并且在小鼠移植物模型中联合使用AZD2811(25 mg/kg,尾静脉静注,每周重复)与维奈妥拉(100 mg/kg、ig、1 次/d)耐受性良好;在体内即使AZD2811 和维奈妥拉间歇给药也能实现持续的抗肿瘤效果[48]。目前小细胞肺癌的治疗主要以化疗联合免疫治疗为主,尚无小分子激酶抑制剂应用到临床。若AURKB 抑制剂研究进展顺利,有望实现该治疗领域零的突破。
1.2 Hesperadin
Hesperadin 是一种基于吲哚啉酮的ATP 竞争性AURKB 抑制剂,IC50为250 nmol/L(无细胞测定法)或3 nmol/L(放射自显影测定法)[49]。Shamsipour等[50]报道Hesperadin会导致异常有丝分裂和细胞分裂受损,用Hesperadin 处理后HeLa 细胞不会增殖(IC50为35~43 nmol/L),并自然成为多倍体。最新的研究发现Hesperidin 在体外和体内均对葡萄膜黑色瘤细胞系(92.1、MEL290、OMM2.3 和XMP46)具有显著的抗肿瘤作用(IC50为 5 nmol/L~7µmol/L),其机制是Hesperidin 损害了端粒酶逆转录酶的启动子组蛋白H3 的磷酸化,从而停止端粒酶逆转录酶的转录[28]。
1.3 SP-96
目前几乎所有开发和研究的AURKB 抑制剂都是ATP 竞争性抑制剂。SP-96 是一种新发现的小分子喹唑啉衍生物,是第1 个针对AURKB 的非ATP竞争性抑制剂[51]。SP-96 具有极高的选择性,对AURKB 的IC50低至0.316 nmol/L。研究表明SP-96可抑制三阴性乳腺癌细胞株MDA-MD-468、肾癌细胞株A498、结肠癌细胞株COLO205 和白血病细胞株CCRF-CEM 的增殖,总细胞生长减少50%的药物浓度(GI50)分别为107、53.2、50.3、47.4 nmol/L[51]。
1.4 西奥罗尼
西奥罗尼是一种强效的ATP 竞争性AURKB 抑制剂,与大多数AURKB 抑制剂一致,西奥罗尼可以抑制AURKB 和组蛋白H3 磷酸化以及诱导G2/M细胞周期阻滞,IC50为9 nmol/L[52]。西奥罗尼也被证明是血管表皮生长因子受体(VEGFR)和集落刺激因子(GSF)-1 受体的有效抑制剂[52]。西奥罗尼是AURKB 特异性抑制剂中唯一的多激酶抑制剂。体内外研究表明西奥罗尼对急性淋巴细胞白血病、非霍奇金淋巴瘤和肝细胞癌均有抑制肿瘤生长作用[52-54]。目前针对西奥罗尼的临床试验已进展至Ⅲ期,感兴趣的靶点主要是晚期实体瘤(小细胞肺癌、卵巢癌、肝细胞癌)和非霍奇金淋巴瘤[55]。
2 进入临床试验阶段的泛AURK 抑制剂
2.1 GSK1070916
GSK1070916 是一种基于氮杂吲哚的ATP 竞争性泛AURK 抑制剂,对AURKB、AURKC 具有高度选择性,IC50分别为0.38、1.5 nmol/L,与AURKA相比,它对AURKB 的选择性高250 倍以上[56],是为数不多对AURKA 几乎没有选择性的泛AURK抑制剂。GSK1070916 已被证明以EC50<10 nmol/L抑制100 多种人肿瘤细胞系肿瘤细胞增殖[57-58]。已有临床试验测试该药在实体瘤的最大耐受剂量为85 mg/m2(静滴、第1~5 天、每21 天重复),剂量限制性毒性为中性粒细胞减少[59]。
2.2 达鲁塞替
达鲁塞替是基于3-氨基吡唑衍生物的泛AURK抑制剂[60],对AURKA、AURKB 和AURKC 的IC50分别为13、79、61 nmol/L(无细胞测定法)[61]。亦有多项体内外试验证实达鲁塞替对多种肿瘤有抑制增殖、诱导凋亡、阻滞细胞周期等作用,如达鲁塞替抑制肝癌细胞系(Hep3B)的细胞增殖24 h IC50为22.03 µmol/L[62-63]。最新的研究发现达鲁塞替可诱导结肠癌细胞凋亡,而BCL2 家族蛋白可拮抗该作用,再次证明抑制BCL2 有助于克服肿瘤细胞对AURKB 抑制剂的耐药性[64]。该药在实体瘤的最大耐受剂量已确认为500 mg/m2(无GSF 支持)或750 mg/m2(有GSF 支持),给药方式为24 h 持续静滴、每14天重复,剂量限制性毒性为中性粒细胞减少[65]。达鲁塞替在淋巴瘤、多发性骨髓瘤的临床试验正在进展中。
2.3 AT9283
AT9283 是一种吡唑-苯并咪唑衍生物,是ATP竞争性泛AURK 抑制剂,对AURKA 和AURKB 表现出相似的选择性,IC50均为3 nmol/L[66],是为数不多对AURKC 几乎没有选择性的泛AURK 抑制剂。AT9283 之前的临床前研究已有系统报道[67]。研究显示AT9283 在酪氨酸激酶抑制剂(TKI)敏感或耐药的慢性粒细胞白血病细胞系中表现出浓度相关(10~100 nmol/L)的抗增殖活性,其机制可能是使细胞周期停滞在G2/M 期以及诱导细胞凋亡[68]。目前已有多项AT9283 应用于非霍奇金瘤、多发性骨髓瘤、难治性白血病和晚期实体瘤的临床试验。
2.4 AMG900
AMG900 是一种基于酞嗪胺的高选择性泛AURK 抑制剂,可竞争性地抑制ATP 与极光激酶活性位点结合,对AURKB 的IC50为4 nmol/L[69]。研究显示AMG900 在体外可通过破坏有丝分裂进程以浓度相关性方式(0.1~100 nmol/L)抑制胶质母细胞瘤细胞系(A172、U-87MG、U-118MG)的生长[70]。AMG900 亦可通过诱导多倍体化和/或凋亡抑制急性髓系白血病细胞生长,与阿糖胞苷联合使用有协同增效作用;在小鼠移植瘤模型中,肿瘤植入后第9 天起使用两种给药方案(22 mg/kg、连续4 d或12.6 mg/kg、连续7 d)口服空载体或AMG900;与空载体组相比,AMG900 显著降低了骨髓中MOLM-13 细胞分数,并且7 d 方案比4 d 方案更大程度地降低了肿瘤负担[71]。与AT9283 相同,AMG900 亦可通过使细胞周期停滞在G2/M 期以及诱导细胞凋亡实现对TKI敏感或耐药的慢性粒细胞白血病细胞系浓度相关(10~500 nmol/L)的抗增殖活性[68]。目前每日口服AMG900 在白血病、实体瘤的最大耐受剂量已确定为25 mg(无GSF 支持)或40 mg(有GSF 支持),剂量限制性毒性为中性粒细胞减少[72]。
2.5 CYC116
CYC116 是嘧啶-2-胺衍生物,也是一种ATP 竞争性泛AURK 抑制剂,对AURKA、AURKB 和AURKC 的IC50分别为19、69、9.2 nmol/L[73]。用1.25 µmol/L CYC-116 处理7 h 可以完全抑制HeLa细胞裂解物中组蛋白H3 磷酸化[74]。1 项CYC116针对晚期实体瘤I 期临床试验已经启动,但赞助商提前终止了试验。体内研究表明,在多种实体瘤和白血病异种移植物模型中,CYC116 都有令人惊艳的抗肿瘤效果[75]。最新的研究表明,CYC116 还可以显著促进干细胞来源的心肌细胞的成熟[76]。
2.6 伊洛拉塞替
伊洛拉塞替也是一种ATP 竞争性泛AURK 抑制剂,对AURKA、AURKB 和AURKC 均表现出强大的抑制作用[77],IC50分别为120、7、1 nmol/L,并且是VEGFR 和AURK 双激酶抑制剂[78]。伊洛拉塞替对多种白血病、淋巴瘤、实体瘤细胞系有抗增殖活性(IC50为0.3~21 nmol/L)[78]。截至目前针对该药物已经进行了4 项临床试验,包括3 项I 期试验和1 项II 期试验,所有这些试验都针对晚期实体瘤进行概念验证和药效学/药动学分析,目前已确立最大耐受剂量为180 mg(口服、1 次/d),最常见的治疗相关不良事件分别为疲劳、厌食和高血压[77]。
2.7 TAK-901
TAK-901 是一种ATP 竞争性泛AURK 抑制剂,对AURKA 和AURKB 的IC50分别为21、15 nmol/L[79],是另一个对AURKC 几乎没有选择性的泛AURK 抑制剂。体外疗效已在多种癌症细胞系中得到证实,IC50=40~500 nmol/L,EC50=50~200 nmol/L[79]。研究发现在胶质母细胞瘤U-87MG 中TAK-901 以剂量相关方式显著降低了细胞生长、活力、自我更新、迁移和侵袭,并诱导细胞凋亡和细胞周期停滞,其机制可能与TAK901 下调胆固醇调节元件结合蛋白1(SREBP1)的表达和激活有关[80]。目前已有2 项针对TAK-901 的I 期临床试验启动。
2.8 BI 847325
BI 847325 是5-烷基吲哚酮衍生物,是选择性丝裂原活化的细胞外信号调节激酶(MEK)和泛AURK 双激酶ATP 竞争性抑制剂[81],对AURKA、AURKB 和AURKC 的IC50分别为25、3、15 nmol/L[82]。体内外模型显示,BI 847325 已在许多细胞系有抗肿瘤效果,并且对BRAF、KRAS 突变阳性的恶性肿瘤最显著[1]。每天以10 mg/kg 的剂量ig给药BI 847325 已被证明在BRAF 和KRAS 突变的小鼠异种移植物模型有效[83]。BI 847325 可通过抑制MEK 克服了BRAF 抑制剂耐药性,这种作用在BRAF 抑制剂获得性耐药模型中得到了进一步的检验和证明[1]。已有研究证实MEK 抑制剂曲美替尼和泛AURK 抑制剂BI-831266 联合使用可有效抑制胰腺导管腺癌小鼠移植物模型的生长[84],因此鉴于BI 847325 是MEK 和泛AURK 双激酶抑制剂,或许BI 847325 单药用于胰腺导管腺癌也能取得理想抗肿瘤活性。最新的研究发现BI 847325 可诱导甲状腺癌细胞凋亡,在体外三维培养模型中观察到,使用BI 847325 处理在分子和/或细胞水平上降低了多药耐药性、细胞周期进展、增殖、血管生成和侵袭[81]。1 项针对BI 847325 用于晚期实体恶性肿瘤的I 期临床试验已经开始。
3 其他泛AURK 抑制剂
还有数个通过临床前验证的泛AURK 抑制剂,有ZM447439、PHA-680632、逆转素(Reversine)、GSK650394、CCT129202、CCT137690、槲皮素、吲哚-2-酮衍生物、LXY18 等,大多数表现出抗肿瘤活性,尚未启动相关临床试验[85-93]。VX-680(MK-0457)、SNS-314、BI 811283、BI 831266、PF-03814735、阿立塞替已进入I 期临床试验阶段,显示出良好的耐受性,并正在推动未来的研究[94-96]。有部分天然产物,如杰多霉素可抑制AURKB,这可能是杰多霉素的抗肿瘤作用机制之一[97-98]。
将AURKB 抑制剂相关临床试验信息进行归纳总结,见表1(来源于ClinicalTrials.gov,数据截至2023 年10 月6 日)。

表1 AURKB 抑制剂临床试验信息Table 1 Clinical trials of AURKB inhibitors
4 结语
极光激酶是细胞分裂过程中保护遗传稳定的重要有丝分裂酶之一。这些酶的异常表达促进了正常细胞向癌细胞的转化。由于AURKB 不可或缺的生理作用,多项针对该靶点的小分子抑制剂被研制出来以期应用到抗肿瘤治疗。近年来AURKB 作为癌症治疗的潜在靶点取得了重大进展,这代表了抗癌药物开发成果鼓舞人心。目前研发的AURKB 抑制剂在体内体外均显示出理想的抗肿瘤效果,AURKB 选择性抑制剂在临床试验中亦表现出良好的前景。随着科学对癌症的进一步深入了解,它们将继续持续改进,抑制AURKB 活性的药物未来应用于临床应该是可行的、可实现的。
最新的研究在53 种不同来源的肿瘤细胞构建的移植瘤模型中发现,对AURKB 抑制剂敏感的肿瘤细胞表现出BH3 相互作用结构域死亡激动剂(BID)高表达的特点,进一步研究发现约6%的实体瘤患者BIDmRNA 高表达[99]。另有1 项治疗则发现伴随AURKA/AURKB 扩增的Burkitt 淋巴瘤患者对传统治疗极度不敏感[100]。肝细胞癌患者中AURKB 的表达与Child-Pugh 分级、微血管侵犯、Edmondson-Steiner 分级和肿瘤复发密切相关[101]。以上研究为如何筛选AURKB 抑制剂临床潜在获益患者提供思路。
发现高选择性、强效和良好药理特性的新型抑制剂是未来的任务。笔者认为针对AURKB 抑制剂的研究可重点关注4 个方向:(1)开发和测试更特异的AURKB 抑制剂;(2)纳米制剂在降低抑制剂
的毒性负荷和提高其功效方面非常有前景,研究应集中在小分子AURKB 抑制剂纳米制剂上;(3)设计临床试验策略时应纳入AURKB 抑制剂与其他具有抗肿瘤活性的小分子抑制剂或传统化疗药物或免疫治疗的联合治疗;(4)通过基因检测筛选潜在获益患者亚群。
利益冲突所有作者均声明不存在利益冲突
