新生儿CHARGE 综合征5 例临床及基因变异分析
2023-11-22王颍源方盼盼陈蒙蒙刘大鹏康文清
王颍源 方盼盼 陈蒙蒙 郭 静 刘大鹏 康文清
郑州大学附属儿童医院 1.新生儿重症监护室,2.郑州市儿童感染与免疫重点实验室(河南郑州 450018)
CHARGE 综合征(OMIM:214800)是一种罕见的先天性常染色体显性遗传疾病[1],发病率约为1/17000~1/15000,30%患者在5岁前死亡[2]。研究发现,CHD 7基因变异是导致CHARGE 综合征的主要遗传学病因[3]。目前国内CHARGE 综合征报道相对较少,新生儿期诊断的报道更少。本研究应用高通量测序技术和Sanger测序技术等对5个新生儿期CHARGE 综合征家系进行基因检查,总结其临床特征、诊治经过、转归,通过家系分析为其提供遗传咨询。
1 对象与方法
1.1 研究对象 收集2018年1月至2020年12月在郑州大学附属儿童医院就诊的5 例CHARGE 综合征患儿样本及家系资料图(图1),男4例、女1例,均为新生儿期发病。5例患儿均经监护人签字同意,接受医学全外显子基因分析及家系验证。本研究经河南省儿童医院伦理委员会审查批准(No.2021-K-076)。

图1 5 例患儿家系图及耳、眼部表现
1.2 方法
1.2.1 诊断依据 依据2016年Hale等[4]提出的CHARGE 综合征的诊断标准,主要指标:①眼部缺损;②鼻后孔闭锁或腭裂;③外耳、中耳或内耳发育异常,包括半规管发育不全;④致病性的CHD7变体。次要指标:①颅神经功能障碍包括听力丧失;②吞咽困难或喂养困难;③脑结构异常;④生长发育迟缓/自闭症;⑤下丘脑-垂体功能异常(包括生长激素和促性腺素不足)或生殖器发育不良;⑥心脏、食管等纵膈气管畸形;⑦肾脏异常,骨骼或四肢发育异常。满足2项主要标准+任意1项次要标准即可诊断CHARGE综合征。
1.2.2 临床资料收集 收集5例患儿的主要表观畸形、临床特征、重要的辅助检查结果等,总结其临床特征及诊治经过。
1.2.3 基因测序 抽取患儿及父母静脉血3 mL,EDTA 抗凝,送至复旦大学附属儿科医院儿科研究所,采用全外显子组基因进行Illumina HiSeq2500高通量测序。去除接头及低质量序列,采用BWA 软件将其定位到人类基因组的参考序列(UCSU 19)上进行比对。针对筛选出的可疑致病位点,用Primer 5.0 设计引物进行扩增及Sanger 测序分析。采用Chromas2进行测序结果分析。
2 结果
2.1 患儿临床特征
5 例患儿均在新生儿期发病,其中4例存在反应差、呼吸困难,均接受机械通气治疗,1 例不明原因发生呼吸心跳骤停,3 例多次撤离呼吸机均失败,1例接受心脏手术,1 例围手术期死亡。5 例患儿均存在不同程度耳部畸形,3 例有眼底病变脉络膜或虹膜缺损。患儿临床特征见图1、表1。
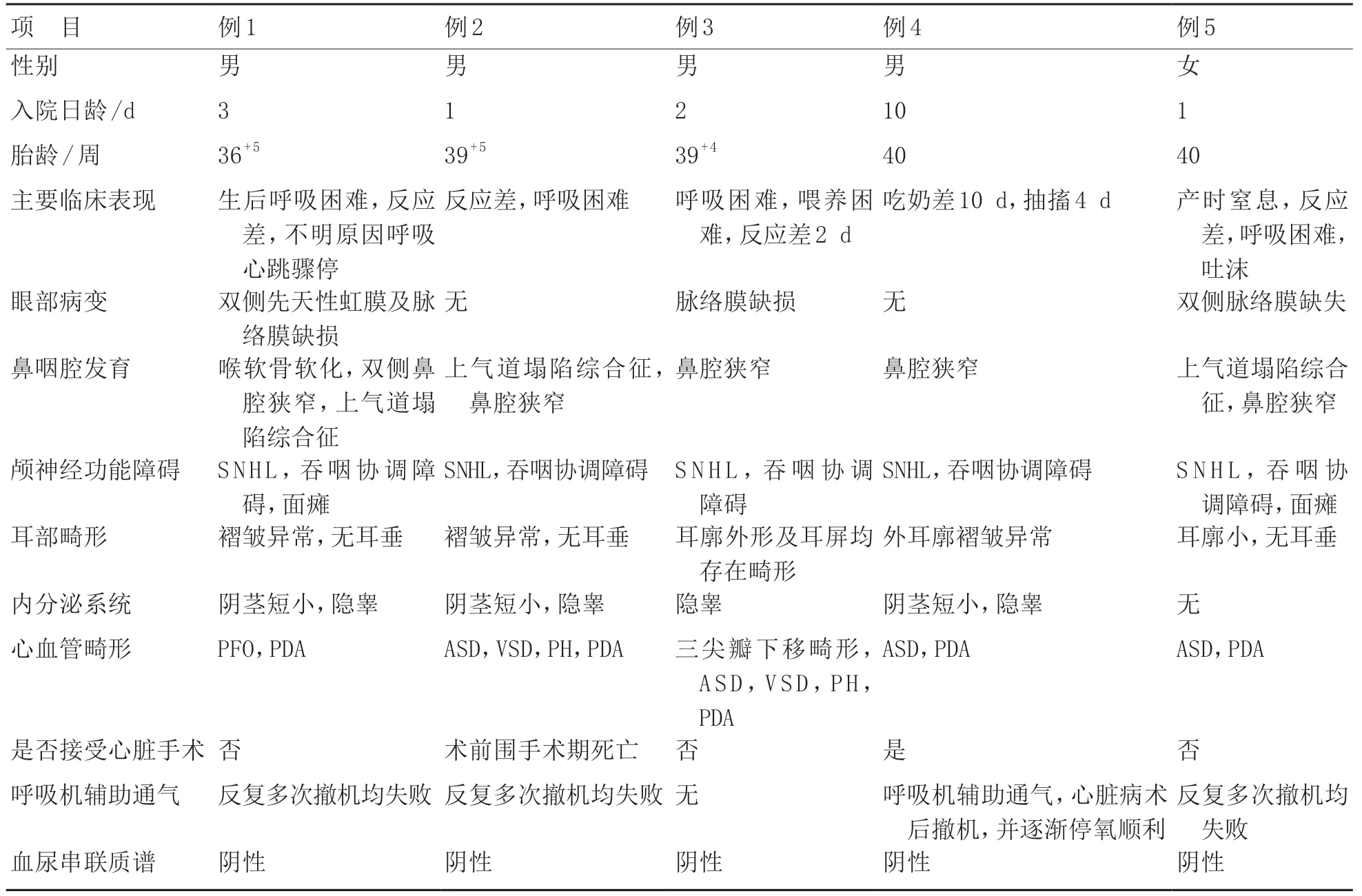
表1 5例新生儿CHARGE综合征患儿临床特征
2.2 基因检测结果
家系1为CHD7基因c.478del(Y160Tfs*51)杂合移码变异,家系2为CHD7基因c.5428C>T(P.R 1810*)杂合无义变异,家系3 为CHD 7基因c.6292 C>T(P.R 2098*)杂合无义变异,家系4 为CHD7基因c.4317delA(p.Q1440S fs*3)杂合移码变异,家系5为CHD7基因c.469C>T(P.R157*)杂合无义变异(表2)。家系4 为未报道变异,其变异经Sanger 测序验证确认(图2)。Sanger 测序显示5个先证者的父母均未发现上述变异。5 例患儿基因变异致病性分析见表3。
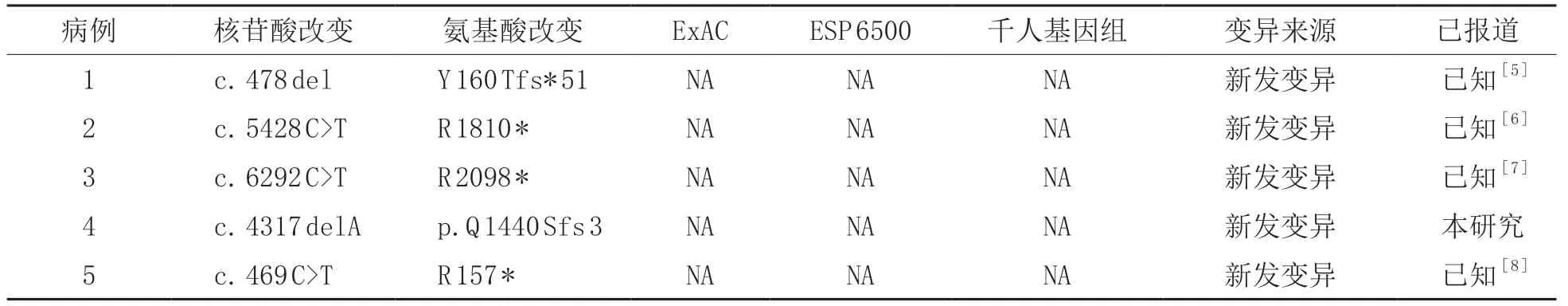
表2 5例新生儿CHARGE综合征患儿CHD7基因变异检测结果
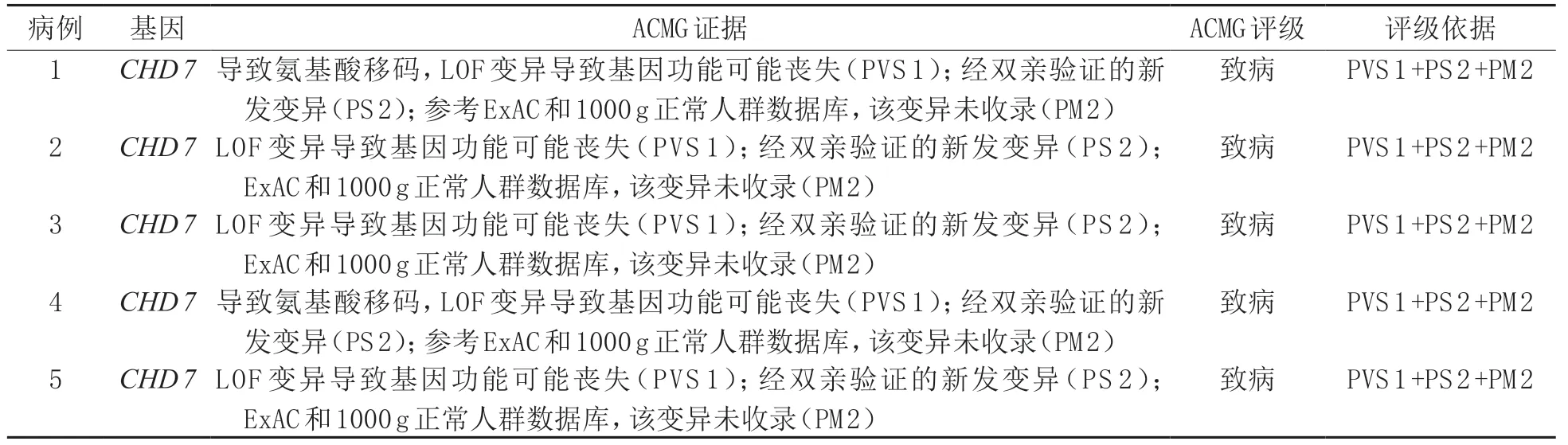
表3 5例患儿基因变异致病性分析

图2 例4 患儿及父母CHD7 基因Sanger 测序结果
2.3 临床转归及预后
病例1、5 因反复撤离呼吸机失败,家属放弃治疗后当天死亡。病例2 因PDA,动脉导管直径大,术前死于感染性休克。病例3 因吞咽障碍,喂养困难,家属放弃治疗后死于家中。病例4 接受心脏手术后控制感染逐渐脱离呼吸机,临床治愈出院,早期曾予以鼻饲奶喂养,情况好转后改口服,现患儿2岁5个月,身高82 cm,体重10 kg(低于同龄同性别正常儿童2 个标准差),目前仍不能独立行走,仅可说“妈妈”。
3 讨论
CHD 7 是哺乳动物细胞中ATP 依赖性染色质域解旋酶DNA 结合蛋白家族的九个成员之一,人类CHD7基因位于8q12.1,基因组全长188 kb,其蛋白包含2 997个氨基酸[4],广泛表达于胚胎和成体多种组织,其变异所产生的影响可存在于整个孕期,尤其是孕早期的3 个月(3~9 周),不仅影响神经系统和生殖系统发育,还影响嗅觉、耳、心脏、骨骼等器官的发育,故CHARGE 综合征患者的表型具有异质性与复杂性[9]。1998年Blake等[10]首先提出本病的诊断标准,认为CHARGE 综合征的诊断应包含4 个主要指标和7个次要指标,主要指标:①眼组织缺损、小眼畸形,②后鼻孔闭锁或狭窄,③耳形态异常或耳聋,④颅神经功能障碍;次要指标:①心脏畸形,②气管食管畸形,③生殖器发育不良或青春期发育迟缓,④唇裂和/或腭裂,⑤发育迟缓,⑥生长迟缓,⑦特征性面容。典型的CHARGE 综合征为符合4 项主要指标或3项主要指标+3项次要指标。后随着研究的深入,CHD 7基因被确定为CHARGE 综合征主要的致病基因[3]。2005年Verloes[11]提出了新的诊断标准,将颅神经功能障碍归于次要指标,同时次要指标去除了“唇裂及腭裂”和“特征性面容”,但由于其具有临床表现多样、病情轻重程度不等、异质性强等特点,漏诊误诊十分常见。2016年Hale等[4]在基因研究的基础上提出了CHARGE 综合征的新的诊断标准,在Verloes诊断标准的基础上主要指标增加了致病性CHD7变体,次要指标上增加了“吞咽困难或喂养困难”和“肾脏异常、骨骼或四肢发育异常”。依据Verloes标准,与儿童及成人相比,在新生儿期典型的CHARGE 综合征临床诊断并无困难,但非典型CHARGE 相应的畸形无特异性,诊断难度较大,可能漏诊一些症状轻或不典型患儿,可疑该病的患儿建议按照诊断标准积极完善相关影像学检查,同时行相关基因检测提高诊断率,避免漏诊,尤其是不完全符合Verloes 标准者,亦不能排除CHARGE 综合征的诊断[12]。按最新的Hale标准,在本组5例患儿明确合并CHD 7基因变异之后,所有患儿均符合CHARGE综合征的诊断。
本研究5 例患儿均为早期新生儿,发病均以多发畸形、呼吸困难为共同特点,且均合并鼻腔狭窄,后者是其呼吸困难的主要病因,考虑该畸形是本组患儿住院治疗效果差的主要原因,但本组无鼻后孔闭锁的病例,所以鼻后孔闭锁可能不是该类患儿必须存在的畸形。另外本研究发现鼻腔狭窄或上气道塌陷等是本组患儿上气道的主要畸形类型。心脏畸形是CHARGE 综合征和具有致病性CHD 7变异患者的主要特征。与非综合征性心脏畸形相比,CHD7致病变异患者更常见PDA 及ASD,本组5 例患儿均有ASD 及PDA[13]。例1 及例5 患儿存在歪嘴哭表现,主要表现为哭闹时口角歪向一侧,上下唇不对称,该体征在新生儿期并不罕见,主要在22q11.2微缺失综合征患者中存在,但多项报道指出歪嘴哭可能为该病的临床表现之一[14-16]。本组患儿均存在不同程度的喂养困难,具体表现为以喂养困难就诊,或临床诊治过程中发现患儿口腔分泌物多,吞咽功能差,与相关研究报道的12 例CHARGE 综合征患儿共同临床表现为喂养困难的结论一致,其提出喂养困难同时合并多发畸形需考虑CHAEGE 综合征的可能[16]。此外例3 在喂养困难的基础上,合并小口畸形,与文献报道的CHARGE 综合征患儿合并T 细胞功能障碍导致原发性免疫缺陷症[17]有关,但本组病例因入院年龄较小均未行相关免疫功能检测。例1、3、5 均合并不同程度眼部畸形。4 例男性患儿合并小阴茎及隐睾,主要是因为CHD 7在胚胎期高表达于嗅球嗅束、垂体前叶、中叶细胞,CHD7变异将导致性激素神经元不能正确迁移以及嗅球发育不良,文献报道大约有60%~80%的CHARGE 综合征患者合并有低促性腺激素及性腺功能减退[18]。新生儿的嗅觉判断尤其是危重新生儿嗅觉判断目前尚无特定方法,故本组患儿不能确定是否合并嗅觉障碍。
消化系统和呼吸系统疾病是CHARGE 综合征生后死亡的主要原因[19]。本组最终4 例患儿死亡,其中例1和例5主要是因住院时间长,反复撤机失败死亡,考虑原因为鼻腔严重狭窄,上气道塌陷,黏膜水肿明显,呼吸道分泌物多,后死于呼吸衰竭;例2亦不能撤离呼吸支持手段,彩色多普勒超声心动图提示动脉导管粗大,准备手术过程中死于严重的围手术期感染;例3患儿虽仅为鼻导管吸氧,存在喂养困难,家属考虑预后可能不良放弃治疗后死于家中,随访过程中发现该患儿死亡原因可能为呼吸困难(衰竭)及喂养不足,最终导致全身衰竭。只有例4临床外观畸形相对较轻,无明显气道发育畸形存在,但需管饲喂养,现生命体征尚平稳,但发育较同龄儿落后。故新生儿合并上气道畸形尤其是诊断CHARGE综合征的患儿要优化呼吸道管理策略,积极防治感染,同时联合眼科、耳鼻喉、口腔等学科尽早介入,开展相应治疗手段早期去除威胁患儿生存的畸形(如合并上气道塌陷综合征的患儿是否可开展相关手术扩大咽腔空间),以提高患儿生存率。
国内有关CHARGE 综合征病例的报道较少。本研究5 例患儿CHD 7基因变异位点中有3 例无义变异、2 例移码变异,变异类型与之前报道基本一致[20]。本组患儿表现出与既往报道患者不同的表型如鼻腔狭窄、上气道塌陷综合征及吞咽或喂养困难等表明,CHARGE 综合征是一种高度异质性疾病,此类患儿易合并其他疾病[21],CHD7基因分析对于全面评估很重要[22]。对于具有CHD 7致病变异的CHARGE 综合征患者,基因型和表型之间的关系尚不清楚[23],文献报道杂合错义变异患者表型相对较轻,无义变异者表型较重,本研究发生无义变异的3例患儿均死亡,另外2例移码变异患儿死亡1例,存活1例,结果与文献报道一致[24]。
综上所述,本研究明确了5 例患儿多发畸形的遗传学病因,并认定CHD 7基因变异为4 例患儿死亡的遗传学病因,丰富了CHD7基因变异的数据库,并为家系遗传咨询提供了重要依据。
