基于加权基因共表达网络分析挖掘茯苓抗食管癌作用靶点
2023-11-16蒋向辉徐玉平
蒋向辉, 徐玉平
(凯里学院大健康学院,贵州 凯里 556011)
食管癌(esophageal cancer,ESCA)是世界上最多见癌症类型之一.根据《2021年全球癌症统计》,它的发病率位居第十,死亡率位居第六[1].食管癌的5年生存率为20%,且因性别而有很大差异[2].根据组织学分类,食管癌可分为食管腺癌(esophageal adenocar cinoma,EAC)和食管鳞癌(esophageal squamous cell carcinoma,ESCC).近十年来,食管腺癌发病率在世界各地均呈现逐年增长的趋势[3],严重危害了人类的生命健康.茯苓为多孔菌科真菌茯苓Poriscocos(Schw.) Wolf的干燥菌核,是传统的药食两用中药,现代药理学研究表明,茯苓具有抗肿瘤、抗肝纤维化、免疫调节、抗过敏等功效[4].研究食管癌的发病机制并挖掘茯苓治疗食管癌的潜在作用靶点将为食管癌的早期筛查和治疗提供重要参考.
加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)是用于探索不同样本之间高度相关基因模块的系统生物学方法[5],通过分析模块与临床性状的关联,挖掘出与临床性状紧密相关的关键模块,经过模块内分析和网络可视化识别模块内的关键基因,这有助于找到与食管癌发生发展相关的核心基因.WGCNA方法已广泛应用于肿瘤和其他疾病的预后标志物和治疗靶点的筛选,特别是在胶质母细胞瘤[6]、前列腺癌[7]等多种癌症当中得到了广泛的应用,效果较好.本研究基于差异表达分析筛选出TCGA-ESCA数据集和GSE22954数据集中的差异表达基因,运用WGCNA方法进行模块化分析找出与食管癌显著相关的基因模块,通过比较两个关键模块中的差异表达基因,获得最终的食管癌相关基因.对食管癌相关基因进行gene ontology (GO) 分析和kyoto encyclopedia of genes and genomes (KEGG) 富集分析,并进行蛋白质相互作用分析将核心基因可视化,进行生存分析对核心基因进行外部验证,利用临床相关性分析来考察关键基因在不同临床分组间的表达差异,借助HPA数据库验证基因在蛋白水平的表达.研究结果表明MCM2、CHAF1A的低表达与食管癌患者的生存期恶化有关,为食管癌的临床诊治开创了新思路.
1 方法
1.1 研究流程
茯苓有效活性成分及其作用靶点筛选→食管癌转录组数据与GEO数据交集基因筛选→交集基因GO和KEGG富集分析→食管癌关键基因筛选→关键基因临床分析→小鼠食管癌模型构建→茯苓水煎液对食管癌关键基因表达影响检测.
1.2 茯苓有效活性成分、对应靶点的检索和候选药物作用靶点的筛选
在TCMSP数据库(https://tcmspw.com/tcmsp.php)中获取茯苓所有的活性成分.根据筛选指标:口服生物利用度(oral bioavailability, OB) ≥ 30%、类药性(drug likeness, DL) ≥ 0.18,得出茯苓的主要活性成分及其对应的靶点[8].利用universal protein (Uniprot)数据库将靶基因全称置换为基因symbol,删除重复项后将基因名输入STRING数据库(https://string-db.org/),构建蛋白互作(protein-protein interaction,PPI)网络图.借助Cytoscape软件筛选出前6个抗食管癌的候选药物作用靶点.
1.3 数据收集与预处理
在TCGA数据库(https://portal.gdc.cancer.gov/)中下载的转录组数据共有171例样本,其中包括160例食管癌和11例正常组织,共19 600个基因.在TCGA数据库中下载的临床数据共183例样本.从GEO数据库(https://www.ncbi.nlm.nih.gov/gds)中下载的GSE22954数据集共有90例样本,其中包括80例正常样本和10例食管癌样本,将基因探针转换为基因名,去重后共获得8 622个基因用于后续分析.
1.4 共表达网络的构建与关键模块的识别
R语言运行WGCNA数据包,将TCGA-ESCA和GSE22954的基因表达数据谱构建成共表达网络,创建基因聚类树状图.参照Miller等[9]验证特定的模块与正常、肿瘤状态的关联性,并绘制模块—性状热图,鉴定出关键模块.其中,相关系数绝对值最大的模块被看作是与临床性状紧密相关的关键模块,将用于后续分析.此外,通过对基因显著性(gene significance,GS)和模块隶属度(module membership,MM)相关性进行计算,以衡量基因与临床性状、基因与各模块特征基因的相关性.
1.5 差异表达分析和交集分析
参照Miao等的方法[10],将limma数据包分别应用于TCGA-ESCA和GSE22954数据集中筛选食管癌组织和正常组织之间的差异表达基因(differentially expressed genes,DEGs).接着运用R软件的ggplot2数据包,将TCGA-ESCA和GSE22954数据集的DEGs可视化为火山图;运行pheatmap数据包绘制DEGs的热图,运行VennDiagram数据包,将关键模块中的基因和差异表达基因取交集,获得最终的食管癌相关基因,并以韦恩图的形式呈现.将最终的食管癌相关基因与茯苓的前6个候选药物作用靶点进行对比,最终确定茯苓抗食管癌的潜在作用靶点.
1.6 GO和KEGG富集分析
参照Yu等[11]的方法利用R软件运行org.Hs.eg.db数据包将基因名转换成基因ID,再利用clusterProfiler、ggplot2和enrichplot数据包对食管癌相关基因进行GO和KEGG富集分析.借助KEGG orthology based annotation system (KOBAS) 数据库(http://kobas.cbi.pku.edu.cn/download.php)中的Gene-list Enrichment功能,检索出基因富集最显著的代谢通路图.
1.7 核心基因的鉴定
参照Szklarczyk等[12]的方法借助STING工具构建食管癌相关基因的PPI网络图,导出PPI数据,Shannon等[13]的方法使用Cytoscape对其进行可视化分析,并进行核心基因的查找.已有研究发现,最大团中心性(maximal clique centrality,MCC)算法是寻找PPI网络中关键节点最有效的方法[14],下载Cytoscape软件的插件CytoHubba,利用它计算每个节点的MCC值并排序.本研究参照张燕玲等[15]的方法,将MCC值位于前十的基因作为核心基因.
1.8 生存分析和临床相关性分析
根据在TCGA数据库中下载的食管癌相关核心基因的表达量,将食管癌患者分成高低表达两组.运行R语言的survival数据包对核心基因进行总生存期(overall surviva, OS) 分析,使用gene expression profiling interactive analysis (GEPIA2) 数据库(http://gepia.cancer-pku.cn/index.html)进行无病生存期(disease free survival, DFS)分析[16],方法选择RFS,按中位数分组,肿瘤名称选择食管癌.若p< 0.05,则说明该核心基因的表达差异对食管癌患者的生存期恶化具有显著影响.提取临床数据中的临床性状,只保留分期列与基因ID列,并按分期进行排序.运行R语言的ggpubr数据包对核心基因进行临床相关性分析,当不同分期之间的p< 0.05,则说明该核心基因在食管癌患者不同分期间的表达差异具有统计学意义.
1.9 免疫组化分析
借助HPA数据库(https://www.proteinatlas.org)获取核心基因在正常样品与食管癌样品中的免疫组化图片,观察基因在正常样品与食管癌样品间是否真的具有差异.
1.10 茯苓水煎液对关键基因表达量影响检测
1.10.1 不同浓度茯苓水煎液的制备 参照Huang等[17]的方法,取茯苓200 g于90 ℃下水煎三次,每次加水300 mL,过滤,合并三次滤液,40 ℃下减压蒸馏,得浸膏18 g,加水90 mL得浓度为0.2 g·mL-1的水提液母液(高浓度),取母液30 mL分别稀释2倍和5倍,制得浓度为0.1 g·mL-1(中浓度)和0.04 g·mL-1(低浓度)的2种茯苓水煎液稀释液.
1.10.2 小鼠食管癌模型构建与关键基因表达量检测 雄性SCID小鼠(20~25 g)后肢皮下注射约6×105个ECA109细胞,40只荷瘤小鼠共分成四组,接种后3周开始灌胃茯苓水煎液,实验组茯苓水煎液浓度分别为0.2 g·mL-1(高浓度)、0.1 g·mL-1(中浓度)和0.04 g·mL-1(低浓度),正常组给等量的水,每日给药1次,连续15 d.采用TRIZOL试剂分离小鼠食管总RNA,将总RNA逆转录成cDNA,采用qRT-PCR方法检测关键基因mRNA表达水平,参照Rubie等[18]的方法以人的磷酸甘露糖酶HMM1基因作为内参基因,qPCR引物序列见表1.

表1 关键基因RT-PCR检测引物
2 结果
2.1 茯苓有效活性成分、对应靶点的检索和候选药物作用靶点的筛选
在TCMSP数据库中检索到茯苓的活性成分共34种,经筛选得15种有效活性成分(见表2):去氢齿孔酸、常春藤皂苷元、茯苓新酸C、茯苓新酸B、茯苓新酸A、茯苓酸、3β-羟基-24-亚甲基-8-羊毛甾-21-酸、多孔覃酸C、过氧化麦角甾醇、星鱼甾醇、去氢土莫酸、啤酒甾醇、去氢茯苓酸、栓菌酸、3β,16α-二羟基羊毛甾-7, 9 (11), 24-三烯-21-酸.其中去氢齿孔酸的OB、DL值最高,故去氢齿孔酸可能是茯苓发挥功效的核心活性成分.

表2 茯苓有效活性成分的相关信息
检索有效活性成分的靶点并去重,最终获得17个作用靶点.按MCC算法从大到小排序,筛选得前6个候选药物作用靶点(图1):CHRM1、NCOA2、PGR、ADRA1B、ADH1B、PTGS2.
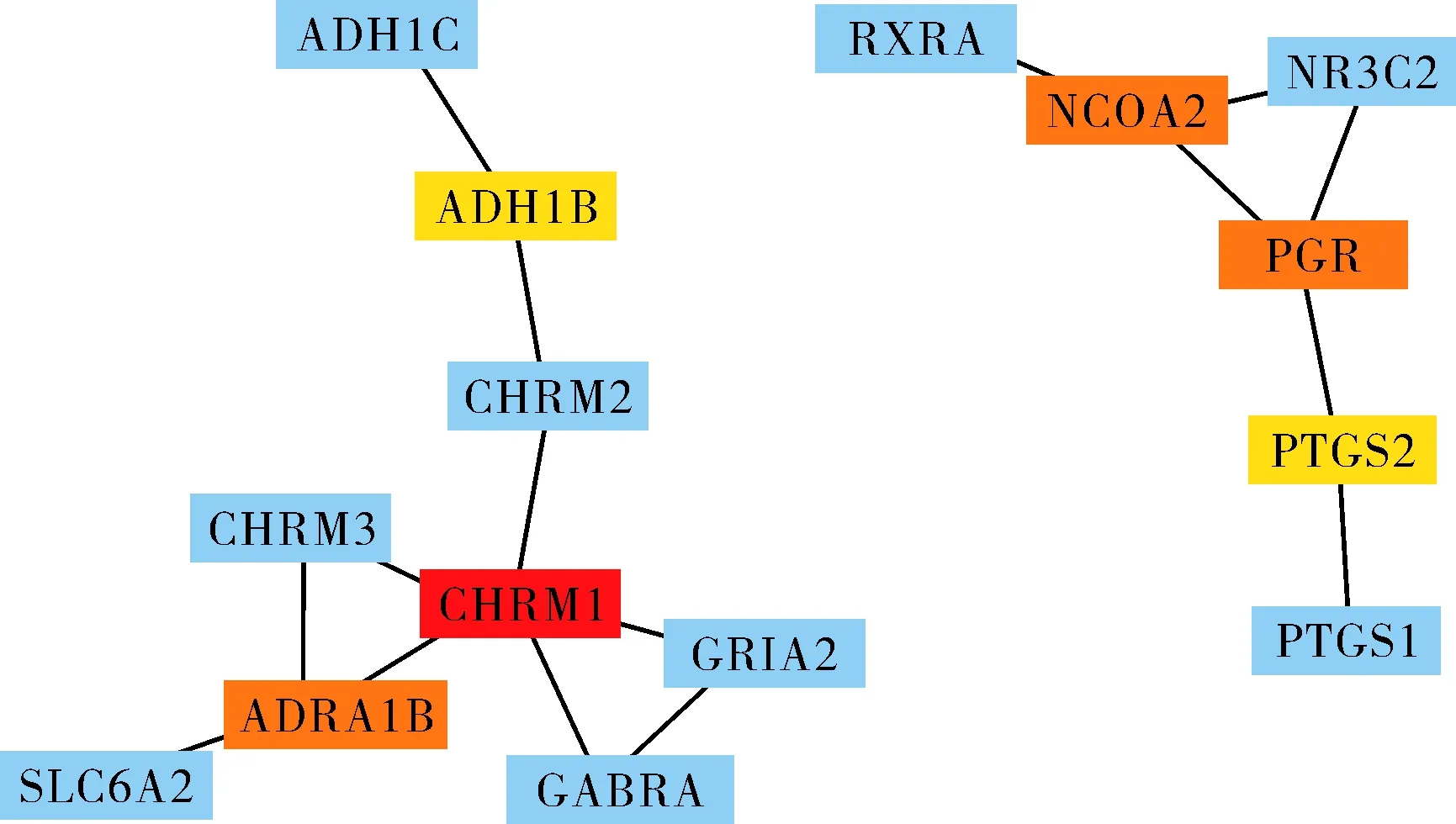
图1 茯苓候选药物作用靶点的鉴别Fig.1 Identification of drug target of Poria cocos
2.2 共表达网络的构建和关键模块的识别
利用WGCNA数据包构建TCGA-ESCA和GSE22954数据集的共表达网络.过滤掉具有缺失值的基因和样本,TCGA-ESCA数据中有171例样本(图2a),GSE22954数据中有90例样本,分别被用于构建样品聚类图(图2b),结果表明两个数据库中的样本均未出现离群样本.

a.基于TCGA-ESCA数据集的样品聚类图; b.基于GSE22954数据集的样品聚类图图2 样品聚类图Fig.2 Sample cluster diagram
为达到无尺度网络标准,需筛选合适的软阈值,利用“pickSoftThreshold”函数计算结果如图3所示,本研究选取了R2大于0.9的β,最终TCGA-ESCA数据集和GSE22954数据集选取的软阈值分别为β=2和7.根据这两个软阈值,分别对两个数据库中的基因作聚类分析,并构建基因聚类树.使用动态切割法进行基因模块的识别,设定每个基因模块中的基因数不少于50,设置MEDissThres的值为0.25,最终TCGA数据库中的基因被分成10个模块(图4a):Black(93个)、Blue(2 722个)、Brown(1 214个)、Green(388个)、Greenyellow(52个)、Magenta(79个)、Pink(91个)、Red(262个)、Turquoise(9 260个)、Yellow(498个).GSE22954数据集中的基因被分成14个模块(图4b):Black(387个)、Blue(1 240个)、Brown(2 709个)、Green(589个)、Greenyellow(143个)、Grey(591个)、Magenta(334个)、Pink(351个)、Purple(222个)、Red(423个)、Salmon(70个)、Yellow(920个).其中Grey模块指难以分配给其余任何模块的基因簇.

a. 基于TCGA-ESCA数据集的软阈值的确定; b.基于GSE22954数据集的软阈值的确定图3 软阈值的确定Fig. 3 Determination of soft threshold
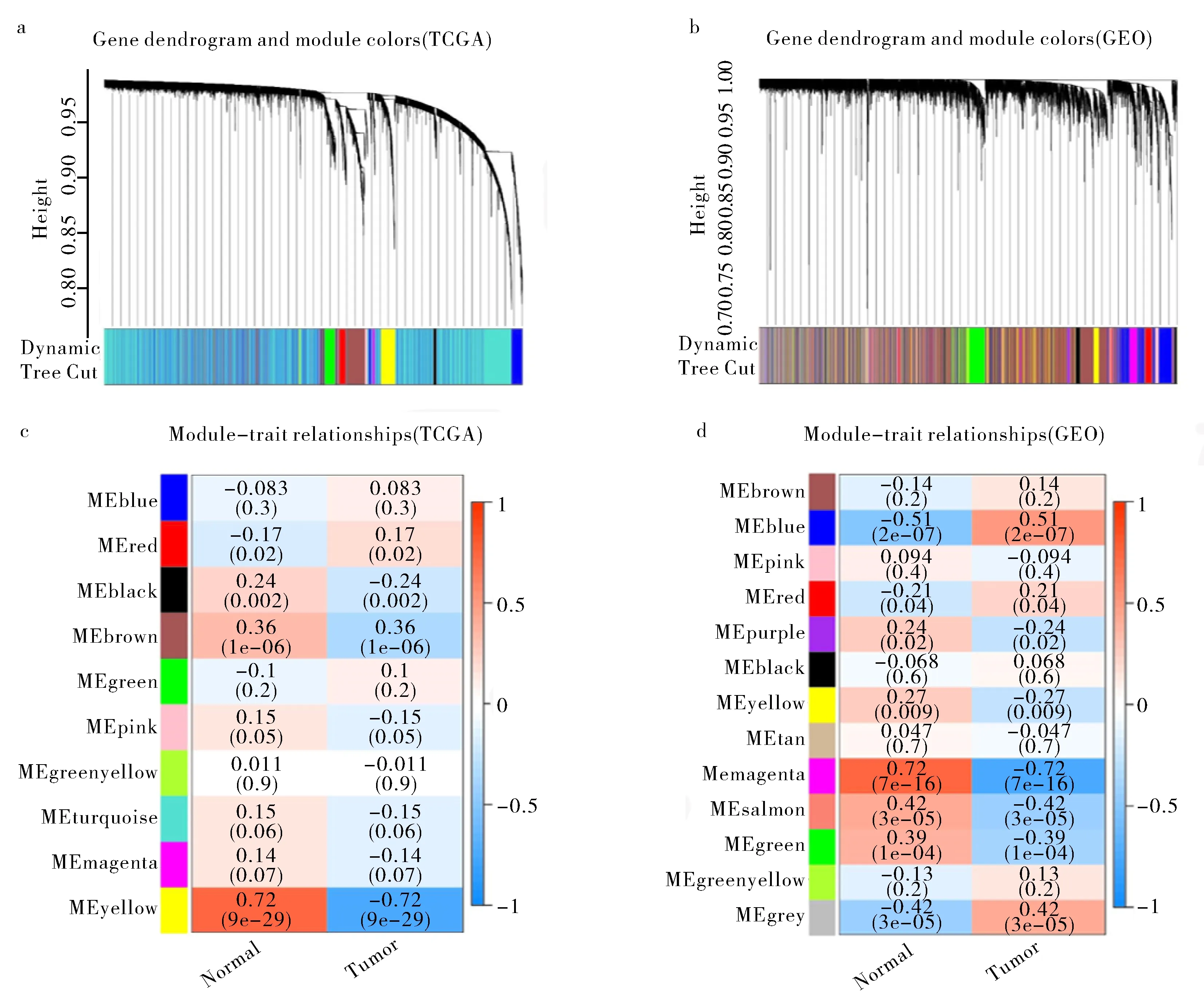
a.TCGA-ESCA数据集的基因聚类树状图; b.GSE22,954数据集的基因聚类树状图;c.TCGA-ESCA数据集的基因模块-特征关系热图; d.GSE22,954数据集的基因模块-特征关系热图图4 聚类分析模块的确定Fig.4 Determination of cluster analysis module
图4c和图4d中每个格子含有两个数值,上方数值代表该模块与临床状态的相关系数,下方数值指相关性检验的p值.在TCGA数据集中,Yellow(r=-0.72,p=9×10-29),Black(r=-0.24,p=0.002),Brown(r=-0.36,p=1×10-6)模块与食管癌组织呈负相关,Red(r=0.17,p=0.02)模块与食管癌组织呈正相关,Pink(r=-0.15,p=-0.05),Blue(r=0.083,p=0.3),Green(r=0.1,p=0.2),Greenyellow(r=-0.011,p=0.9),Magenta(r=-0.14,p=0.07)Turquoise(r=-0.15,p=0.06)模块同临床性状的相关性较弱.在GEO数据集中,Blue(r=0.51,p=2×10-7),Red(r=0.21,p=0.04)模块与食管癌组织呈正相关,Green(r=-0.39,p=1×10-4),Magenta(r=-0.72,p=7×10-16),Salmon(r=-0.42,p=3×10-5),Purple(r=-0.24,p=0.02),Yellow(r=-0.27,p=0.009)与食管癌组织呈负相关.Black(r=0.058,p=0.6),Brown(r=0.14,p=0.2),Greenyellow(r=0.13,p=0.2),Pink(r=-0.094,p=0.4)模块与临床状态的相关性较弱.结果表明TCGA-ESCA中的Yellow模块和GSE22,954中的Magenta模块与食管癌组织的相关性最高.进一步对Yellow模块(cor=0.87,p=1.9×10-15)和Magenta模块(cor=0.670,p=7.2×10-45)内的GS和MM相关性进行计算,也验证了这两个模块为关键模块(图5).
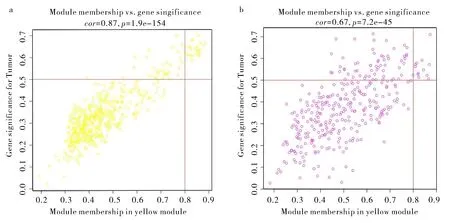
a. Yellow模块的GS与MM相关性; b.Magenta模块的GS与MM相关性图5 GS与MM相关性Fig. 5 Correlation between GS and MM
2.3 差异表达分析和取交集基因
利用limma数据包对TCGA-ESCA(图6a)和GSE22954(图6b)数据集中的基因进行了差异表达分析,得到差异热图.其中横坐标表示样品(浅蓝色为正常样品、浅红色为食管癌样品),纵坐标表示基因,若某基因在正常样品中是红色,表示它在正常样品中高表达,而在食管癌样品中有多种颜色,则表明它在食管癌样品中是低表达.在火山图中,横轴上的值表示log FC,纵轴上的值表示校正后的p值.黑点指的是某基因在正常样品和食管癌样品中无表达差异,绿点指的是某基因在食管癌样品中下调,红点指的是某基因在食管癌样品中上调.

a. TCGA-ESCA数据集差异基因热图; b.GSE22954数据集差异基因热图图6 差异热图和火山图Fig.6 Differential heat map and volcano map
与正常食管组织相比,TCGA数据集中共鉴定出2 394个差异表达基因在食管癌组织中表达失调,其中1 225个下调,1 169个上调.GSE22954数据集中共鉴定出1 505个差异表达基因在食管癌组织中表达失调,其中872个下调,595个上调.通过火山图(图6)和热图(图7)可视化.提取TCGA数据集中黄色模块和GSE22954数据集中品红色模块的共表达基因,并与前面所得的差异表达基因进行交集分析,最终识别出29个交集基因,结果通过韦恩图可视化(图8).将交集基因与茯苓抗食管癌的6个候选作用靶点作对比,发现ADH1B可能为茯苓抗食管癌的潜在作用靶点.

a. TCGA-ESCA数据集差异基因的火山图; b.GSE22954数据集差异基因的火山图图7 差异热图和火山图Fig.7 Differential heat map and volcano map

图8 交集基因与差异基因Venn图Fig.8 Venn diagram of intersection genes and differential genes
2.4 GO和KEGG富集分析
GO富集分析结果如图9所示,29个交集基因主要富集在细胞核分裂、有丝分裂细胞周期相关的调控、细胞周期G2/M期的转换、姐妹染色单体分离、细胞器分裂等321个生物学过程(biological process,BP),微管结合、微管蛋白结合、3′→5′脱氧核糖核酸解旋酶活性、三磷酸腺苷酶活性等48个分子功能(molecular function,MF),纺锤体、微管、驱动蛋白复合物、中间体等53个细胞组分(cell component,CC).KEGG富集分析结果如图10所示,29个交集基因富集在细胞周期、范可尼贫血途径、氮素代谢、孕酮介导的卵母细胞成熟、DNA复制、细胞衰老这6条代谢通路上.

a. GO富集气泡图;b. KEGG富集柱状图图9 交集基因的 GO和KEGG富集分析图Fig.9 GO and KEGG enrichment analysis of the intersection genes
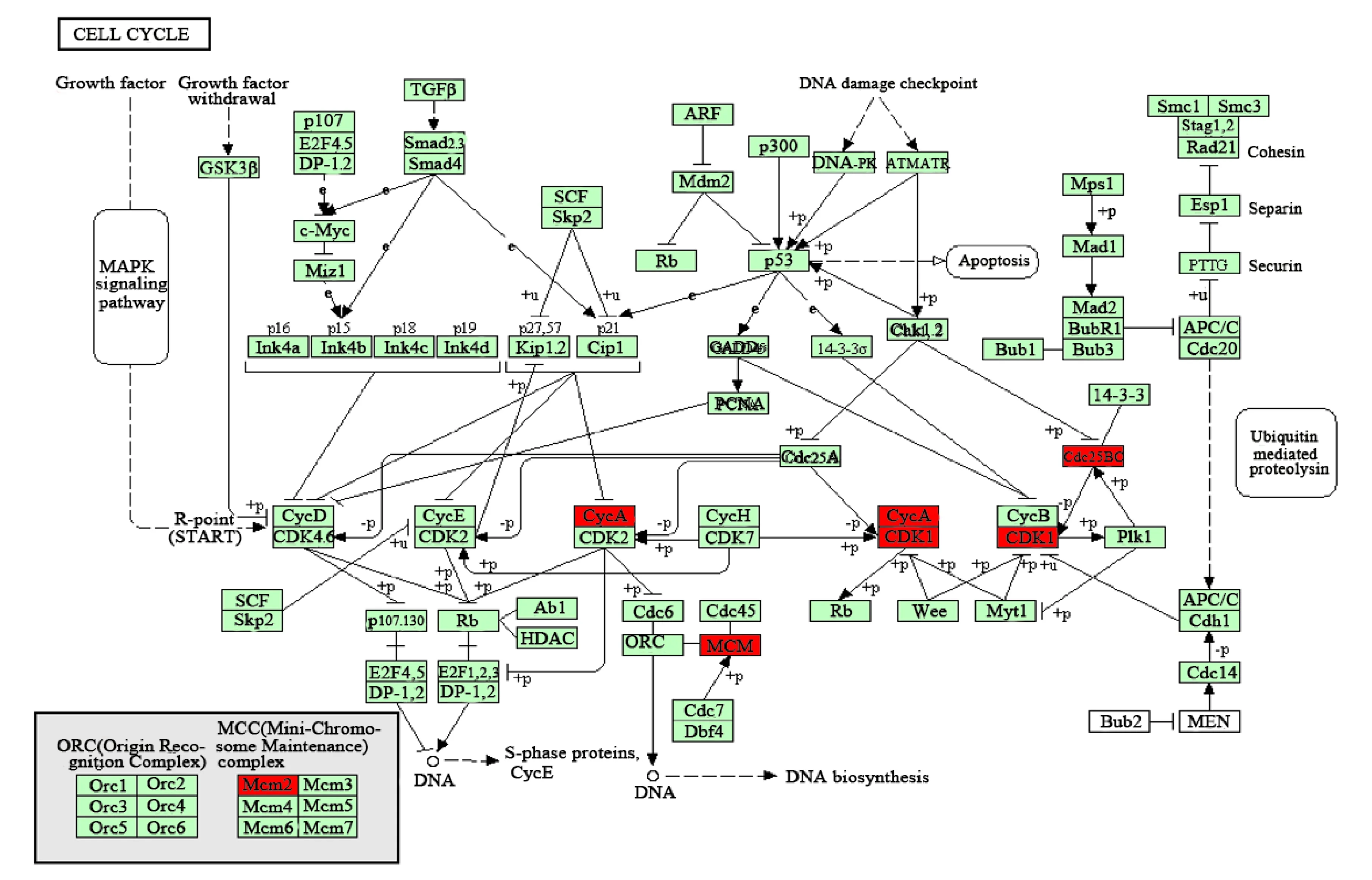
注:红色节点表示食管癌相关基因.图10 代谢通路图Fig. 10 Metabolic pathway diagram
借助KOABS数据库获取了交集基因富集最显著的细胞周期通路图.如图10所示,CDKs是该通路中重要的调控酶,不同的CDK结合不同的周期蛋白(cyclin),在细胞周期不同阶段使各自的底物蛋白磷酸化,改变其结构和功能,从而调节细胞周期各阶段的进程.在G1期,CDK2、CDK4、CDK6将调节因子Rb磷酸化,使HDAC、转录因子E2F复合体得以解离,解离后的E2F在c-myc等物质的帮助下与DNA启动子上的E2F位点结合,启动转录,进一步合成DNA复制所需要的蛋白质.在S期,食管癌相关蛋白MCM被CDC7/DBF4复合体磷酸化,磷酸化的MCM与ORC组成复合体,启动DNA的复制,因此MCM在DNA复制过程中起着重要的作用.在G2期,周期蛋白cyclinA/cyclinB含量积累达到最大值,它们与食管癌相关蛋白CDK1结合形成复合物,Wee1等激酶对CDK1多个特定位点上的氨基酸残基磷酸化.随后在食管癌相关蛋白CDC25B,CDC25C的作用下,去掉某些位点的磷酸基团,此时CDK1开始出现活性,具有活性的食管癌相关蛋白CDK1可以催化多种底物蛋白磷酸化,由此调控细胞从G2期向M期转换.例如催化组蛋白H1磷酸化,促使染色质凝缩;催化核仁蛋白磷酸化,促进核仁解体;催化C-abl蛋白磷酸化,促使细胞形态调整等.
2.5 核心基因的鉴定
利用STING数据库构建29个交集基因的蛋白质互作网络图,图中显示有18个节点和82根边缘线(图11a).运用Cytoscape中的CytoHubba插件计算每个节点的MCC值,按MCC值的大小进行排序,可从蛋白互作网络图中鉴定出前10个核心基因.结果如图11b所示,筛选出的核心基因包括DLGAP5、MCM2、CCNA2、CDK1、TPX2、TRIP13、KIF23、KIF2C、CENPF和CHAF1A.另外,网络图中节点颜色越红,则表明该节点越有可能是网络的核心.
2.6 生存分析、临床相关性分析和免疫组化分析
筛选出前十个核心基因和茯苓抗食管癌的潜在作用靶点ADH1B后,基于TCGA-ESCA的临床数据,运用R软件运行survival数据包作OS分析,结果如图12所示:DLGAP5(p=0.147)、CCNA2(p=0.613)、CDK1(p=0.111)、TPX2(p=0.977)、TRIP13(p=0.647)、KIF23(p=0.702)、KIF2C(p=0.947)、CENPF(p=0.384)、CHAF1A(p=0.334)、ADH1B(p=0.579)的表达量与食管癌患者的OS相关性低,而MCM2(p=

图12 核心基因的总体生存分析图Fig.12 Overall survival analysis diagram of hub genes
0.003)的低表达与食管癌患者的OS恶化显著相关.利用在线工具GEPIA2数据库进行DFS分析,结果如图13所示,CCNA2的结果为logrankp=0.19,HR=1.4,CDK1的结果为logrankp=0.71,HR=0.91,DLGAP5的结果为logrankp=0.15,HR=1.4,TPX2的结果为logrankp=0.21,HR=1.4,TRIP13的结果为logrankp=0.6,HR=1.1,KIF23的结果为logrankp=0.56,HR=0.87,KIF2C的结果为logrankp=0.24,HR=1.3,CENPF的结果为logrankp=0.37,HR=1.2,MCM2的结果为logrankp=0.87,HR=0.96,CHAF1A的结果为logrankp=0.049,HR=0.62.ADH1B的结果为由logrankp=0.56,HR=0.86,由上可知CHAF1A的低表达水平与食管癌患者的DFS恶化显著相关.

图13 核心基因的无病生存分析图Fig.13 Disease-free survival analysis diagram of hub genes
利用R语言的ggpubr数据包对11个基因进行临床相关性分析,结果如图14所示,CDK1、CHAF1A、DLGAP5、TPX2和TPIP13的表达量在Ⅰ期与Ⅱ期间的差异显著性p值依次为0.044,0.039,0.040,0.014,0.035,MCM2的表达量在Ⅱ期与Ⅲ期间的差异显著性p值为0.017,TPX2在Ⅰ期与IV期间的p值为0.032,均小于0.05.因此CDK1、CHAF1A、DLGAP5、TPX2和TPIP13在Ⅰ期与Ⅱ期之间、MCM2在Ⅱ期与Ⅲ期之间、TPX2在Ⅰ期与IV期的食管癌患者之间的表达量是有显著差异的,而其它各期之间的p值均大于0.05,因此基因表达量无显著差异.
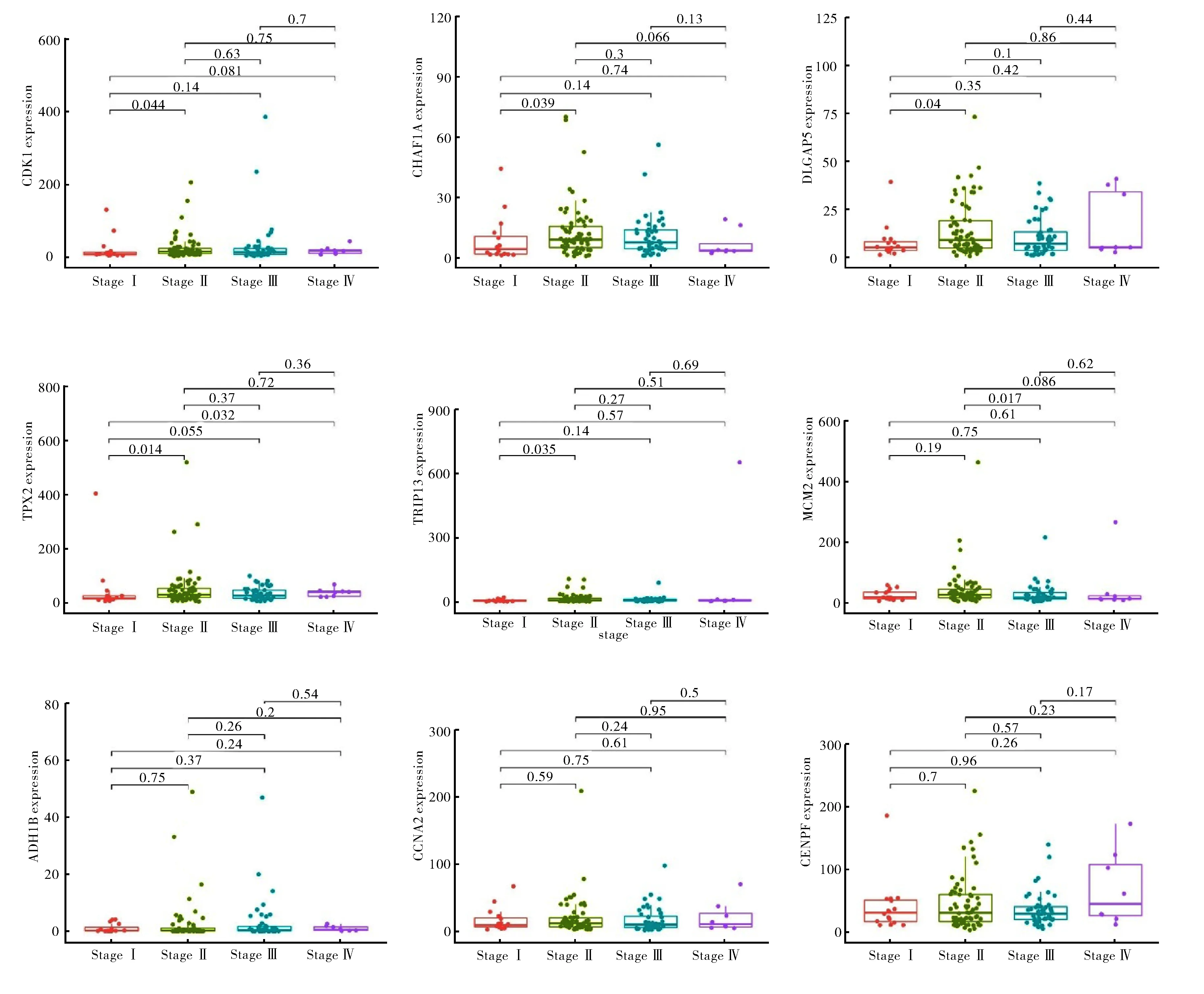
图14 临床相关性分析图Fig.14 Clinical correlation analysis
利用HPA数据库获取MCM2基因在正常组织(图15a)和食管癌组织(图15b)中的免疫组化图片,图像显示MCM2在正常组织中呈现低等强度染色,染色细胞占比小于25%,在食管癌组织中呈现中等强度染色,染色细胞占比为25%~75%.这表明MCM2的表达在食管癌组织中显著上调.
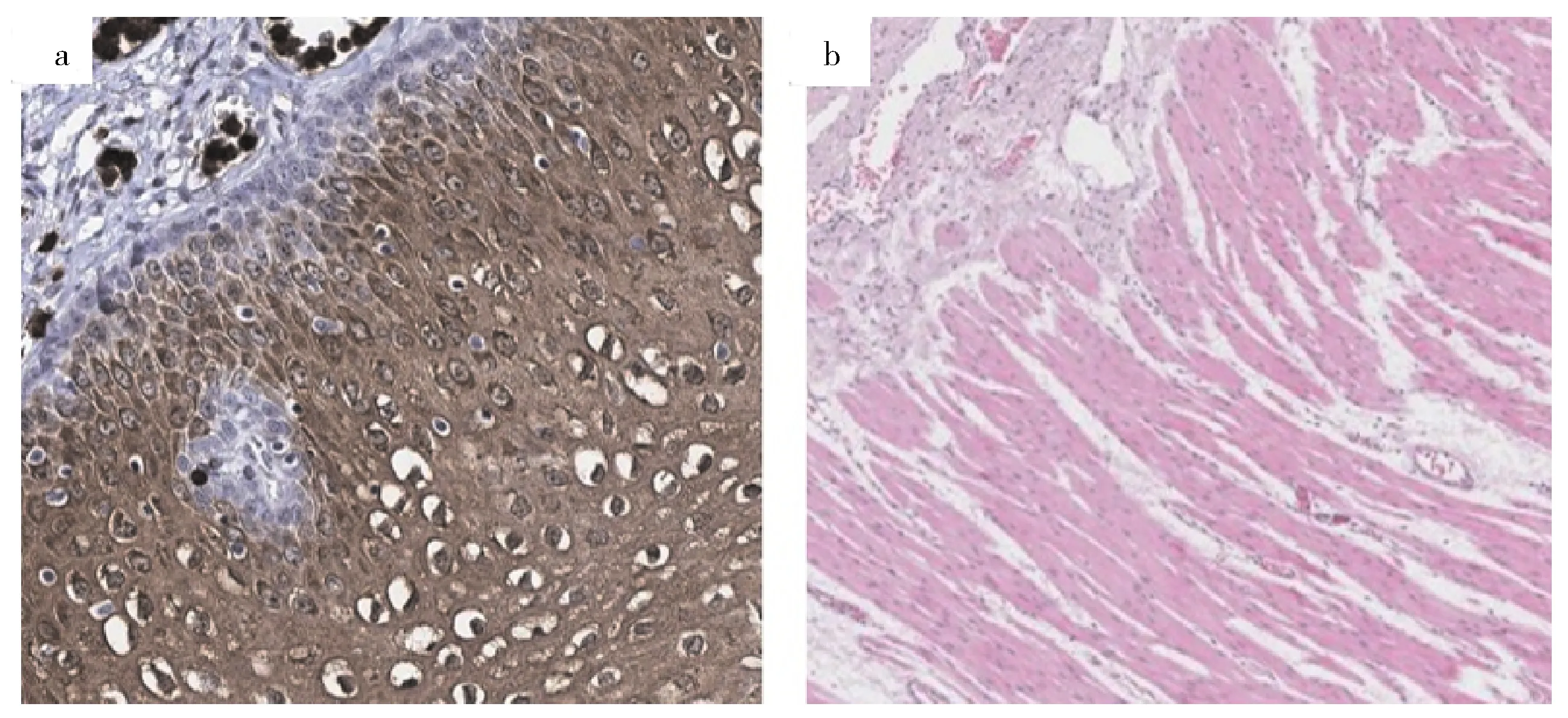
a. 正常组织; b.食管癌组织图15 正常组织与食管癌组织的免疫组化图Fig.15 Immunohistochemical images of normal tissue and esophageal cancer tissue
2.7 茯苓水煎液对关键基因表达量影响
对筛选到的CDK1、CHAF1A、DLGAP5、TPX2、TPIP13和MCM2等6个候选靶点基因进行RT-PCR定量表达分析,结果如图16所示,CDK1、CHAF1A、TPIP13和MCM2基因经不同浓度的茯苓水煎液处理后,表达量与对照存在显著差异(p< 0.05),其中CDK1、CHAF1A和TPIP13经高浓度的茯苓水煎液处理后表达量显著增加,而DLGAP5和TPX2表达量变化差异不明显,但MCM2经不同浓度的茯苓水煎液处理后表达量随着茯苓水煎液浓度先升高后降低.结果进一步显示CDK1、CHAF1A、TPIP13和MCM2是茯苓水煎液治疗食管癌的主要作用靶点.

注:不同小写字母表示差异达到显著水平(p< 0.05),相同字母则表示无统计学差异.图16 茯苓水煎液对关键基因表达量影响Fig.16 Effect aqueous extract from Poria cocos to key gene expression
3 小结与讨论
本研究利用TCMSP数据库检索出茯苓的有效活性成分,其中OB、DL值最高的是去氢齿孔酸,故去氢齿孔酸可能是茯苓发挥功效的核心活性成分.另外对有效活性成分对应的作用靶点进行研究,共获得其作用靶点17个,按MCC排序,最终筛选出6个候选药物作用靶点:CHRM1、NCOA2、PGR、ADRA1B、ADH1B和PTGS2,其中ADHIB被认为是茯苓抗食管癌的潜在作用靶点.ADH1B是乙醇脱氢酶,已有研究表明,ADH1B基因的多态性与食管癌的发病风险有关,ADH1B在乙醇转化为乙醛的代谢途径中起着关键作用,携带ADH1BAA基因型的人,其编码的
乙醇脱氢酶活性较高,乙醇较容易被转换为乙醛.研究发现,茯苓可能是通过其有效活性成分增加ADH1B的活性,从而增强人体抵抗力,抑制食管癌肿瘤细胞的生长.
本研究基于差异表达分析和WGCNA探讨食管癌发生发展相关的核心基因.通过对差异基因进行GO富集分析发现,这些基因主要参与细胞核分裂、有丝分裂细胞周期的G2/M期转换、细胞器分裂等生物学过程,主要与3′→5′脱氧核糖核酸解旋酶活性、微管结合、单链DNA结合等分子功能有关,主要影响有丝分裂纺锤体、微管、驱动蛋白复合物等细胞组分.KEGG富集分析结果表明,29个食管癌相关基因富集最显著的通路是细胞周期代谢通路,在该通路中,食管癌相关基因MCM2是S期内启动DNA复制的重要因子,CDK1在G2/M期转换起着重要的调控作用,它们通过控制细胞周期运转,防止细胞增殖失控,甚至细胞癌变.本研究根据Cytoscape中的插件CytoHubba计算每个节点的MCC值,筛选出前10个核心基因:DLGAP5、MCM2、CCNA2、CDK1、TPX2、TRIP13、KIF23、KIF2C、CENPF和CHAF1A,其中MCM2的低表达与食管癌患者的总生存期恶化显著相关;DFS分析发现CHAF1A的低表达与食管癌患者的无病生存期恶化显著相关.同时,临床相关性分析发现CDK1、CHAF1A、DLGAP5、TPX2和TPIP13表达量在Ⅰ期与Ⅱ期的食管癌患者之间的表达量存在显著差异.CHAF1A是染色体组装因子,它能够调控DNA修复和细胞增殖[19].李雪玲等[20]研究发现,CHAF1A在多种肿瘤中均高表达,其中包括食管癌,并可能是影响肺癌、肝细胞癌和乳腺癌预后的治疗靶点.MCM2是微小染色体维持蛋白2,位于细胞核当中,具有调控DNA复制的功能.目前它已被视作S期内部检验点激活的重要因子、特异性增殖相关因子, 且被认为是癌前标记[21].本研究分别对TCGA-ESCA和GSE22954数据集进行差异表达分析,分析结果均显示CHAF1A和MCM2在食管癌样品中显著上调;生存分析结果显示,CHAF1A和MCM2的低表达水平与食管癌患者的预后不良有关;临床相关性分析提示MCM2从Ⅱ期到Ⅲ期的表达量显著下降,而CHAF1A从Ⅰ期到Ⅱ期表达量显著上升.Zhong等[22]通过食管鳞癌GEO数据库和TCGA数据库比对发现MCM2 与总体生存无相关性.因此MCM2和CHAF1A可能是影响食管癌预后的治疗靶点,同时也是影响食管癌分期的靶点.本研究结果可能为食管癌的早期筛查与靶向治疗提供重要参考.
