木质素磺酸钠衍生S/N共掺杂电催化剂结构调控与OER性能研究
2023-11-14王晓菲薛李静周海潮林绪亮邱学青
王晓菲,薛李静,周海潮,林绪亮,3,邱学青
(1.化学与精细化工广东省实验室 揭阳分中心, 广东 揭阳 515200;2.广东工业大学 轻工化工学院, 广东 广州 510006;3.广东工业大学 广东省植物资源生物炼制重点实验室, 广东 广州 510006)
人类社会对化石能源的过度依赖引发的环境污染和能源危机逐年严重,在全球引起了广泛的关注。氢能作为一种高能量密度的清洁能源,是未来可持续能源体系中最具潜力的载体之一[1-2]。而电解水分解是氢气的重要途径,该过程可以分为析氧反应和析氢反应两个半反应[3-4]。其中,析氧反应涉及四电子转移过程(4OH-→ O2+ 2H2O + 4e-),导致动力学缓慢、能量效率低等问题,是制约整个水电解装置效率的关键因素。因此合理设计催化剂活性结构降低OER反应能垒,是发展电解水制氢产业的关键。贵金属RuO2和IrO2是目前公认的综合性能最佳的OER电催化剂,但其低储量和高成本阻碍了其广泛的商业应用[3,5]。高效稳定的非贵金属基OER电催化剂的开发与研究备受关注。
过渡金属硫化物(Transition Metal Chalcogenides,TMCs)成本低、储量丰富,尤其是独特的物理化学性质、电子性能使其成为一种极具潜力的OER电催化剂[3]。在近年来开发的金属硫化物OER催化剂中,Ni基和Co基硫化物催化剂表现出优异的活性而被广泛研究[6]。然而,过渡金属硫化物催化剂存在活性结构单一、结构不稳定等问题。针对此类问题,近年来研究者通过原子掺杂、构筑高熵金属硫化物等手段对硫化物的电荷状态与中间体吸附强度进行调控,有效提高了OER催化活性[7-9]。金属元素掺杂能够强化催化剂内部电子传输,而非金属元素的掺杂通常可以调节活性位点与中间体之间的吸附。Wang等[10]针对金属体相掺杂和非金属表面掺杂对催化剂结构与表面催化活性的影响机制进行了研究,通过金属Cr 和非金属 P 在 Co3S4体相和表面的双重掺杂,有效调控了催化剂的体相与表面电子结构,增强了催化剂的表面反应活性。然而,由于金属硫化物在OER强氧化条件下,催化剂表面的成分、物相及结构均易发生变化,其结构稳定性的提高依然面临重大挑战[11-12]。采用结构可控的载体材料,强化载体对金属活性结构的结合与分散作用,是稳定金属活性结构的重要手段之一。
引入有机配体与金属配位是有效稳定高分散金属位点的有效方法,然而传统有机配体通常面临环境不友好、成本高等问题[13]。作为自然界储量丰富、唯一的可再生三维芳香结构的生物质原料,木质素被认为是一种理想的碳材料前驱体[14]。木质素磺酸盐(Lignosulfonates, LS)是亚硫酸盐制浆的主要副产物,含有丰富的磺酸基和酚羟基,其磺酸基团与酚羟基均是与金属离子配位的有效位点,是最具潜力的含硫碳前驱体。此外,通过表面改性方法在木质素磺酸盐结构中引入新的官能团,如酰胺类配位基团,不仅可以将氮原子掺入木质素衍生碳骨架中调控电子结构,也可以利用酰胺基团的强配位作用将金属离子嵌在碳骨架中,在与木质素中固有的功能基团的共同作用下,提高金属物种的分散度,进而提升催化活性。
本文通过氧化氨解手段,向木质素磺酸钠中引入适量的酰胺基团,有效提高其对金属的配位能力。在高温碳化过程中一步获得S/N共掺杂的碳纳米片负载Co9S8-Ni3S2协同结构催化剂。结合全面表征对改性木质素络合金属前驱体在热解碳化过程中的结构演变机理进行了深入探索,并对得到的系列催化剂的结构、电化学性质与OER活性进行了研究。高分散Co9S8-Ni3S2的协同结构与N对碳载体的缺陷掺杂有效调控了催化剂表面电子结构,促进高活性位点的暴露。Co9S8-Ni3S2/SN-C在OER反应中表现出较低的过电位和良好的稳定性。本文为木质素磺酸钠应用于新型高效电催化材料提供了理论依据。
1 实验部分
1.1 木质素磺酸钠的氧化氨解改性
木质素磺酸钠的氧化氨解改性步骤如下:将10 g质量分数为25%的氨水溶液加入160 mL的超纯水中,然后将20 g的LS溶解在上述溶液中,并加入30 g 30%的过氧化氢,将混合物在室温下搅拌15 min。然后,将该混合物转移到水热反应器中,在120 ℃下反应90 min,反应结束后,混合物被冷却到室温。将反应产物旋转蒸发除去未反应的反应物和水,真空干燥并研磨成粉末得到催化剂,标记为LS-NH2-x,x表示H2O2和LS的质量比,LS-NH2表示氧化氨解改性后的LS。
1.2 催化剂的制备
取1.0 g LS-NH2溶解在去离子水中得到溶液A,随后称取一定量的Co(NO3)2·6H2O与NiCl2·6H2O溶解在去离子水中得到溶液B,将溶液B在持续搅拌作用下逐滴加入溶液A中,搅拌均匀,测量混合溶液的pH。使用氨水调节混合溶液的pH至9,静置12 h,去除上清液,得到产物记作Co/Ni+LS-NH2pH,冷冻干燥3 d,研磨后即得到木质素-金属复合材料(Co/Ni-LSNH2)。在N2氛围下高温煅烧,自然冷却至室温,使用稀盐酸和超纯水洗涤,60℃干燥12 h,即得到Co9S8-Ni3S2/SN-C催化剂。
1.3 催化剂表征
采用FT-IR光谱仪(美国Thermo Fisher,iS50R)测试有机物含有的官能团,测试波数范围为4 000 ~400 cm-1,分辨率4 cm-1。采用X射线衍射仪(Rigaku MiniFlex-600,Cu Kα(λ= 0.154 1 nm))测试样品的晶体结构,电压35 kV,电流25 mA。采用场发射扫描电子显微镜(SEM,Zeiss Sigma 500,10 kV)测试样品的表面形貌与微观结构。采用透射电子显微镜(TEM,FEI Tecnai G2 f20 s-twin,200 kV)测试观察样品结构。采用X射线光电子能谱(XPS,Thermo KAlpha+)测试分析催化剂表面电子状态与化学结构。
1.4 电化学测试
所有电化学测试均在三电极系统中进行,使用Gamry Interface 1 010电化学工作站。分别用石墨棒和Hg/HgO作为对电极和参比电极。采用“滴涂法”制作工作电极,将4 mg的碳材料粉末加入200 μL 0.5%Nafion-乙醇溶液中,待粉末超声分散均匀后取50 μL滴加在处理过的泡沫镍上,然后用电极夹夹住碳纸作为工作电极。所得催化剂负载量为4 mg·cm-2。
通过线性伏安法(Linear Sweep Voltammetry,LSV)测量极化曲线以表征复合材料的OER催化活性。电解液为1 mol·L-1KOH溶液,极化曲线扫描速率为2 mV·s-1。在LSV测试之前,对样品进行几个循环扫描以获得稳定状态。所有极化曲线数据没有经过iR校正,所有电位均根据可逆性氢电极(Reversible Hydrogen Electrode, RHE)计算得到,公式为
在1.60 V的恒定电位、100 kHz ~ 0.1 Hz 的频率范围下检测得到电化学阻抗谱图(Electrochemical Impedance Spectroscopy, EIS)。通过简单的循环伏安法,在不同扫描速率下由非法拉第区域的双层电化学电容测试电极的电化学表面积(Electrochemical Surface Area, ECSA)。
2 实验结果及讨论
2.1 催化剂结构演变机理分析
通过二维异核单量子相干核磁共振方法(Heteronuclear Single Quantum Coherence-Nuclear Magnetic Resonance, HSQC-NMR)分析了LS和氧化氨解木质素磺酸钠(LS-NH2)结构的主要信息,包括各单元间连接类型、芳香结构和改性结构的分布(见图1)。LS和LS-NH2主要呈现β-O-4、β-β΄和β-5键的特征信号[15]。在这些化学键中,β-O-4键占较高的含量比例,符合常规工业木质素的结构特征。LS-NH2的二维HSQC核磁共振光谱与LS的明显不同,部分的β-5、ββ΄和β-O-4键被氧化断裂(见图1(b)、(d)),并且出现了丰富的与金属配位强度较强的苯甲酰胺衍生物(GBa)结构,有利于提高催化剂活性位点的分散度。

图1 LS和LS-NH2(溶剂:DMSO-d6)的部分2D HSQC NMR光谱(等高线以颜色编码来表示对应的结构)Fig.1 Partial 2D HSQC NMR spectra of lignosulfonates (in DMSO-d6) before and after oxidative ammonolysis modification (Contours are color-coded to the structures responsible)
Co/Ni-LS-NH2复合物是通过将LS-NH2与Co、Ni金属离子配位形成的。如图2所示,LS、LS-NH2和Co/Ni-LS-NH2的FTIR光谱均在1 044 cm-1处出现一个对应于S=O对称拉伸振动的特征峰[16]。与LS相比,LS-NH2和Co/Ni-LS-NH2的FTIR谱图在1 406 cm-1处出现了一个新的特征峰[17],对应于C–N结构,进一步说明LS通过氧化氨解成功引入氮元素。
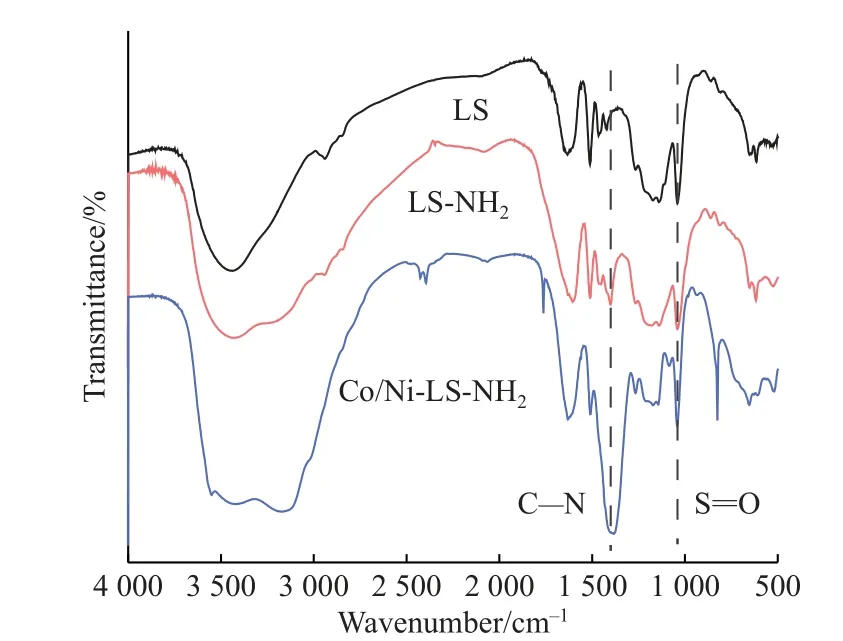
图2 LS、LS-NH2和Co/Ni-LS-NH2复合物的FTIR光谱图Fig.2 FTIR spectra of LS, LS-NH2 and Co/Ni-LS-NH2 complexes
Co/Ni+LS-NH2pH、LS-NH2pH、Co/Ni pH分别是Co/Ni+LS-NH2、LS-NH2、Co/Ni溶液调节pH静置12 h后的上清液,其紫外-可见(Ultraviolet Visible, UVVis)吸收光谱如图3所示。相比于LS-NH2,调节pH后的LS-NH2pH在λ≈ 280 nm的紫外-可见吸收光谱中强度没有明显变化,说明pH的变化对LS-NH2结构没有明显影响。相比于Co/Ni溶液,调试pH后的Co/Ni pH溶液在λ≈ 280 nm紫外-可见吸收光谱中强度没有明显变化,但溶液出现沉淀物,说明调节pH后,金属离子形成氢氧化物沉淀。相比于Co/Ni+LS-NH2溶液,Co/Ni+LS-NH2pH溶液在λ≈ 280 nm的紫外-可见吸收光谱中强度显著变弱,并且出现分层现象,表明LS-NH2与金属离子在调节pH过程中配位形成络合物。如图3(b)所示,相比于Co/Ni+LS-NH2pH,在Co/Ni pH溶液中加入相应质量的LS-NH2粉末后的溶液(Co/Ni pH + LS-NH2)在λ≈ 280 nm的紫外-可见吸收光谱中强度显著增强,进一步证明了催化剂形成过程中LS-NH2与金属离子配位形成络合物。
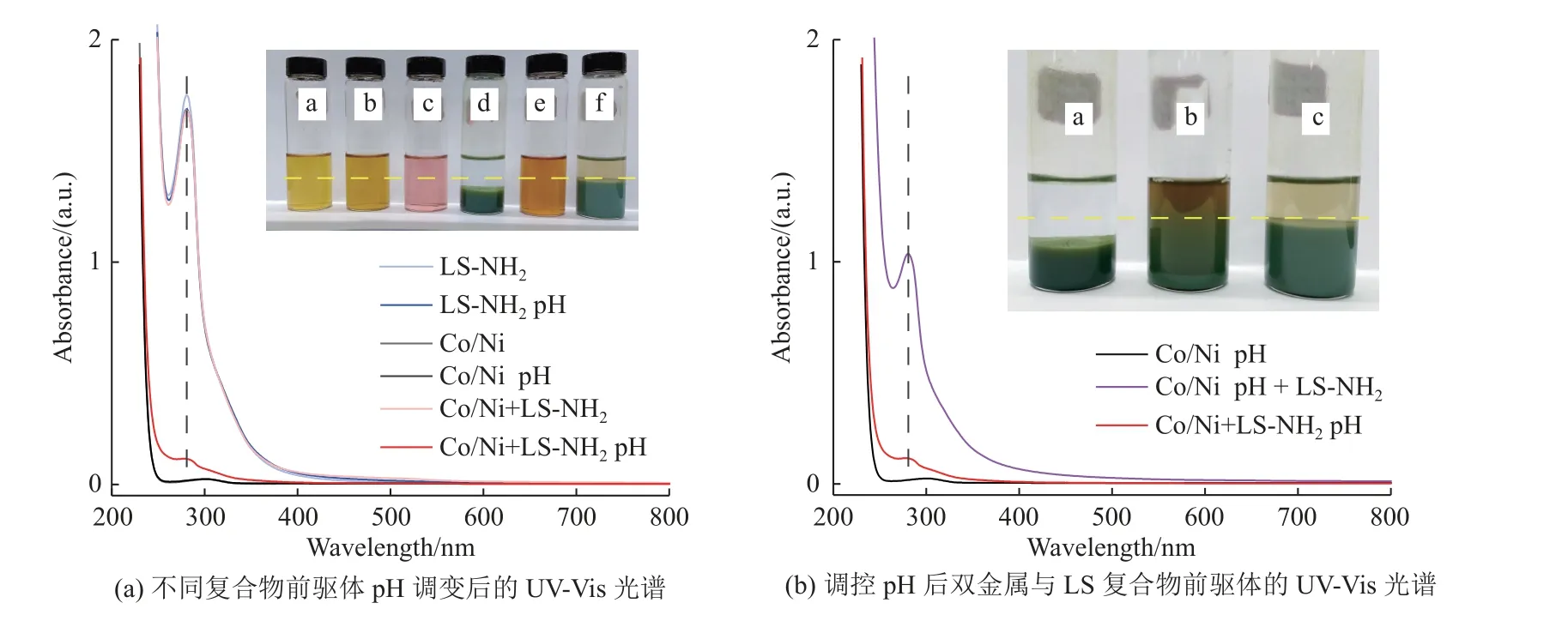
图3 不同前驱体溶液的UV-Vis光谱Fig.3 UV-Vis absorption spectra of different compounds precursor
图4为LS-NH2和M-LS-NH2(M=Ni2+、Co2+)的XPS谱图。LS-NH2的O 1s谱图在533.70 eV、532.09 eV和530.62 eV处呈现3个特征峰,分别归属于–SO3、C–O–C/C–O–H、O=C–N/–COOH。对于M-LSNH2来说,O-M的表面相分布量对于LS-NH2显著增加,表明LS-NH2和金属离子的配位是在简单混合的过程中形成的。LS-NH2的N 1s的XPS精细谱(见图4(b))可以拟合为2个峰,分别对应O=C–N和C–N,表明LS的氧化氨解成功引入酰胺基团。相比于LS-NH2,M-LS-NH2在405.99 eV和399.97 eV处显示出新峰,分别对应–NO3和N–M,表明金属与N配位成功。对于S 2p的XPS精细谱(见图4(c)),LS-NH2的原始曲线被拟合为两个峰,对应两种不同类型的硫物种:和R–,这是木质素磺酸钠的典型结构。相比于LSNH2,M-LS-NH2在160.92 eV处显示出新的峰(S–M),表明金属与S配位成功。以上结果证明了改性后的LS与金属成功配位,有利于提高金属离子(Co、Ni)与改性后的LS之间的相互作用。
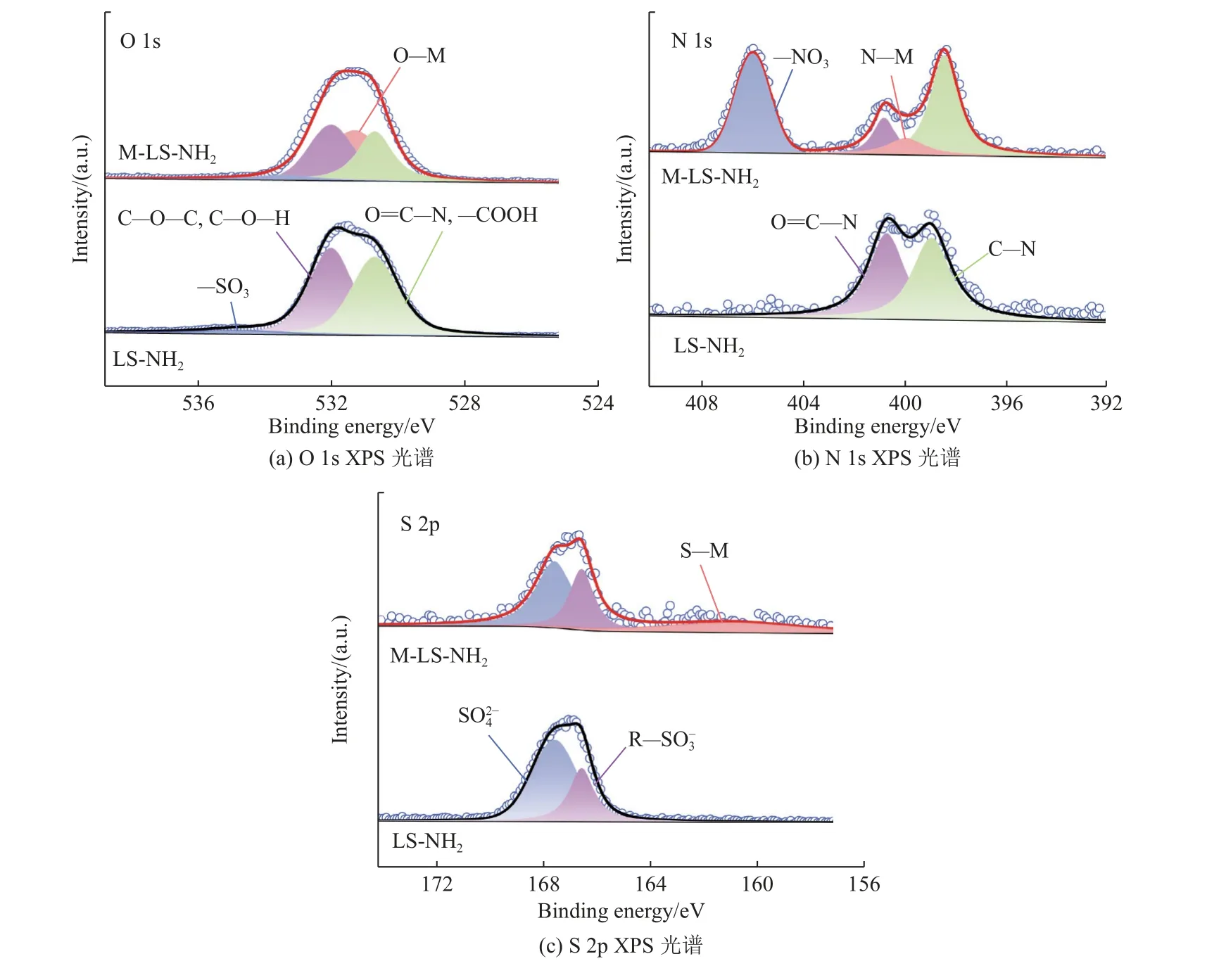
图4 干燥的LS-NH2和M-LS-NH2(M=(Co,Ni))的XPS光谱图Fig.4 XPS spectra of dried LS-NH2 and M-LS-NH2 (M=(Co, Ni) )
基于以上结构分析,总结出Co9S8-Ni3S2/SN-C催化剂的制备过程如图5所示,木质素磺酸钠通过氧化氨解改性反应引入适量酰胺基团作为络合位点,与金属(Co2+和Ni2+)配位形成络合物前驱体,经热解形成具有优异分散性的S/N共掺杂碳负载Co9S8-Ni3S2催化剂。

图5 Co9S8-Ni3S2/SN-C的合成过程示意图Fig.5 Schematic diagram of the synthesis process of Co9S8-Ni3S2/SN-C
采用热重分析(Thermogravimetry, TG)研究LSNH2和Co/Ni-LS-NH2复合物在高温处理过程中的热解情况。如图6(a)~(b)所示,失重过程主要分为3个阶段:常温~ 250 ℃、250 ~ 600 ℃和600 ~ 1 000 ℃。在第一阶段中,LS-NH2和Co/Ni-LS-NH2复合物的失重是由于水分和挥发物的消除。与LS-NH2相比,Co/Ni-LS-NH2复合物的失重明显较高,主要由于部分未与LS-NH2配位的金属盐热解。此外,Co/Ni-LS-NH2复合物的失重速率峰由286 ℃移至228 ℃,表明LS-NH2与Co和Ni金属离子形成的复合物促进了木质素的热解。在第二阶段,LS-NH2和Co/Ni-LS-NH2复合物的失重归因于其众多的含氧基团的分解,形成初级无定形碳结构,释放出芳香烃(如苯酚、愈创木酚或丁香酚)、烷基(如CH4、C2H4或C2H6)、CO2、CO以及一些含有硫和金属的小分子[18]。同时,金属盐在该阶段发生分解与还原。第三阶段,碳材料深度碳化失活。与LSNH2相比,Co/Ni-LS-NH2复合物在367和544 ℃出现新的失重速率峰,处于725 ℃的失重速率峰向高温偏移至830 ℃,表明改性后的LS中的部分Na被Co和Ni金属原位取代。由于钴、镍化合物与含硫官能团之间的活化反应,Co/Ni-LS-NH2复合物经热解过程形成金属硫化物[19-20]。
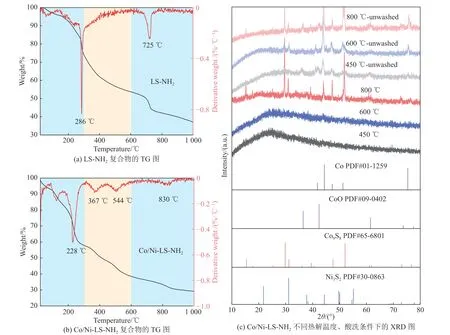
图6 不同催化剂前驱体热重与XRD图Fig.6 TG and DTG curves, XRD patterns of different catalyst precursor
图6(c)为通过XRD表征碳化后Co/Ni-LS-NH2复合物的晶相结构。经450 ℃碳化后,酸洗前催化剂呈现明显的CoO(JCPDS NO.09-0402)、Co(JCPDS NO.01-1 259)和Co9S8(JCPDS NO.65-6 801)衍射峰,而经高温600 ℃碳化后,CoO衍射峰几乎消失。稀酸洗涤后, 经450 ℃和600 ℃高温碳化得到的催化剂上未观察到明显的金属物相衍射峰,而经800 ℃更高温度焙烧后观察到明显的Co与Co9S8晶相结构。表明酸洗后去除了催化剂表面团聚的大尺寸金属颗粒,并且促进了金属在催化剂上的高分散。而焙烧温度的升高能够促进金属的还原,温度过高导致严重团聚现象。
结合上述结果与文献分析[18-21],Co/Ni-LS-NH2复合物在热解碳化过程中可能发生的反应如式(1)~式(7)所示。随着温度的升高可能会生成CoO和NiO等金属氧化物作为中间物质(反应1、2),并进一步与木质素反应形成金属态Co和Ni(反应3、4)。同时,氧化钴的硫化导致Co9S8物相的形成(反应7)。当热解碳化温度升高到800 ℃时,小分子可以相互作用促使金属物相重排形成Co9S8、Ni3S2等(反应6、7)。因此,Co9S8-Ni3S2/SN-C催化剂表现出明显的Ni3S2衍射峰(JCPDS NO.30-0863)和Co9S8衍射峰(JCPDS NO.65-6 801)。
式中:Lignin代表LS-NH2的主体结构。
2.2 催化剂结构演变机理分析
如图7(a)~(b)的SEM图所示,Co9S8-Ni3S2/SN-C催化剂表面存在大量交错的纳米片,并且具有较多相互连通的大孔隙,可能是由热解过程中释放气体导致。这种多孔结构有利于传质过程,提高电化学性能。从图7(c)中观察到纳米颗粒均匀分布在纳米片上,结合图7(d)的统计结果显示纳米颗粒的平均尺寸为6.3 nm,较小的纳米颗粒有助于提供更多的催化活性位点。Co9S8-Ni3S2/SN-C催化剂的SAED图(见图7(e))清晰地展现出两种不同的衍射环。多晶环(白环)与Co9S8的(220)、(422)和(600)面匹配,而衍射斑(黄色圆圈)则分别归属于Ni3S2的(021)和(113)面。HRTEM图像(见图7(f))也证实了这些纳米粒子对应于Ni3S2和Co9S8纳米粒子,其中,晶面间距为0.234 nm的晶相归属于Ni3S2的(021)平面,而晶面间距为0.227 nm的晶相对应于Co9S8的(331)平面。图7(g)中EDS图表明Co、Ni、S和N原子均匀一致地分布在碳层上,证明了它们之间的强相互作用。以上结果表明氧化氨解提高了LS与金属Co、Ni间的配位能力,从而构建了金属分散性高且作用紧密的高效催化剂。

图7 Co9S8-Ni3S2/SN-C催化剂。(a)~(b) SEM图;(c) TEM图;(d)颗粒直径尺寸分布图;(e) SAED图;(f) HRTEM图;(g)EDS Mapping图Fig.7 (a)~(b) SEM image; (c) TEM image; (d) Size distribution diagram of particle diameters; (e) SAED pattern; (f)HRTEM image; (g) EDS Mapping of Co9S8-Ni3S2/SN-C catalyst
通过XPS进一步分析了Co9S8-Ni3S2/SN-C的表面化学结构与价态分布。如图8(a)中XPS广谱所示, C、N、S、Ni和Co元素共存于催化剂表面。图8(b)中Co 2p谱图分别展现了780.0,795.7,781.8,797.4 eV处的特征峰,归属于Co3+和Co2+[22-23],2个卫星峰位于785.3 eV和802.0 eV处。图8(c)中Ni 2p XPS谱图显示Ni2+(855.5,873.0 eV)与Ni3+(856.7,874.2 eV)共存,861.7和879.7 eV处对应卫星峰[24-26]。XRD、HRTEM和XPS结果证明在Co9S8-Ni3S2/SN-C中形成了Co9S8和Ni3S2相。如图8(d)所示,通过对C 1s谱图进行分峰拟合,确定了C–S、C–N结构的存在,证明S、N成功掺入碳层[22,27]。而由S 2p的XPS谱图分峰结果(见图8(e))可知,催化剂中S以3种不同结构存在。位于161.1和162.4 eV处的特征峰归属于S2-(2p3/2和2p1/2轨道),验证了金属硫化物的存在;163.6 eV和167.99 eV的特征峰分别对应噻吩–S结构(C–S–C)和氧化硫,表明S元素被成功引入到载体碳骨架中[28]。如图8 (f)所示Co9S8-Ni3S2/SN-C催化剂的N 1s 的XPS谱图可以拟合为4个峰,分别位于398.4,399.7,401.1和403 eV,对应吡啶–N、吡咯–N、石墨–N和氧化–N,不同N结构的共存与相互转化能够有效调控表面电子结构,促进中间物种的吸附与活化,改善OER反应活性[29]。
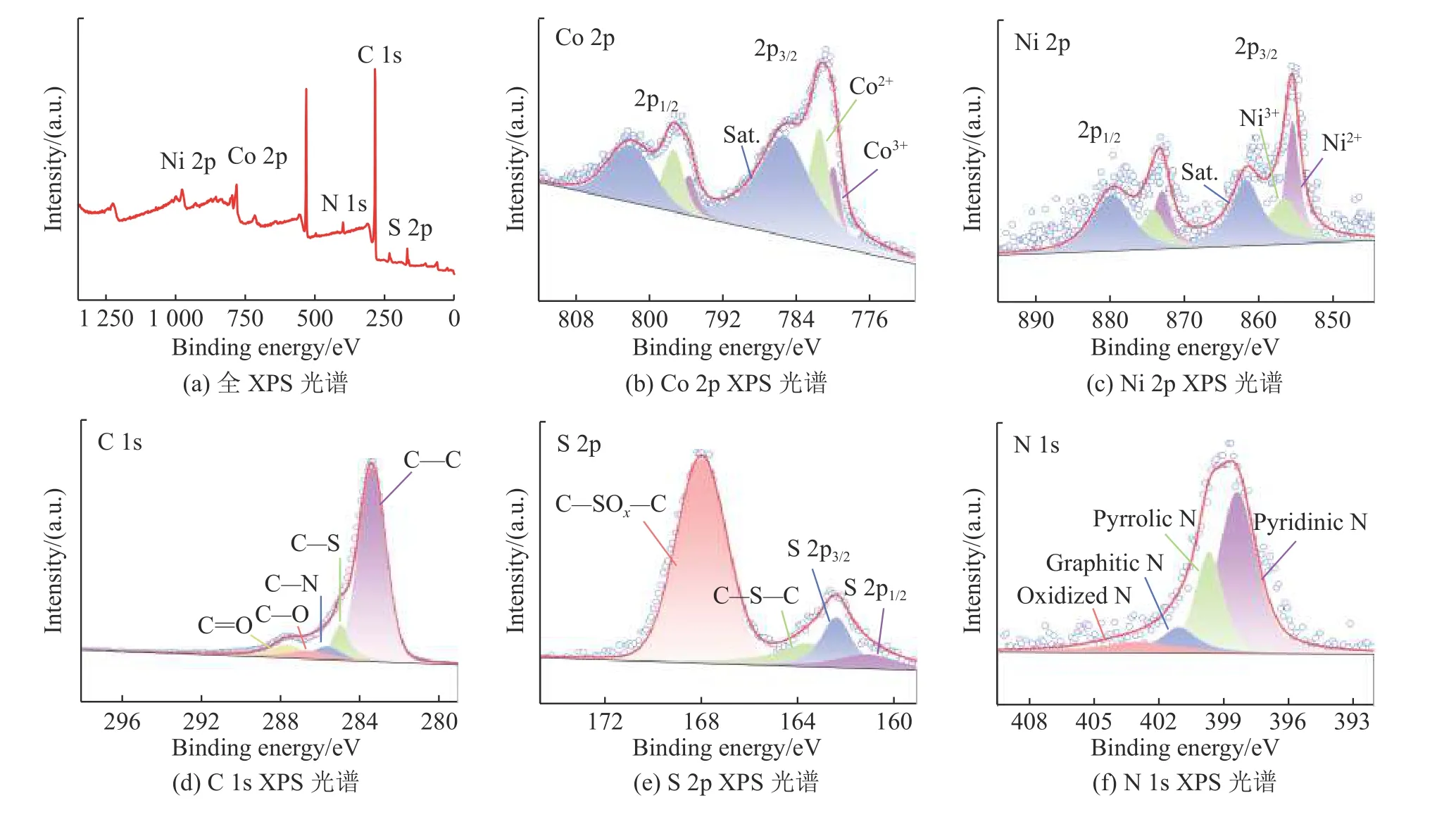
图8 Co9S8-Ni3S2/SN-C的XPS光谱图Fig.8 XPS spectrum of Co9S8-Ni3S2/SN-C
2.3 催化剂的OER性能分析
在1 mol·L-1KOH电解液的三电极体系下,考察了不同氧化氨解程度的木质素磺酸钠制备的系列Co9S8-Ni3S2/SN-C催化剂的OER电催化性能。如图9(a)所示,电流密度达到50 mA·cm-2时,氧化氨解过程中H2O2与LS比例x= 1.5的催化剂过电位为350 mV,优于x为0,1,2和2.5(过电位分别为390,410,380,390 mV) ,说明经过氧化氨解改性后制备的催化剂电化学性能提升,归因于表面杂原子掺杂对能带结构与电子环境的有效调控。如图9(b)所示,x= 1.5对应的Tafel斜率为171.5 mV·dec-1,低于x= 1 (176.mV·dec-1)和x= 2.5 (190.7 mV·dec-1),高于x= 0 (139.5 mV·dec-1)和x= 2 (140.4 mV·dec-1),表现出较好的OER电催化性能。此外,通过测量该系列催化剂的ECSA(见图9(c))发现,当x= 1.5时,催化剂的Cdl为2.16 mF·cm-2,大于x= 0 (0.31 mF·cm-2)、x= 1 (0.62 mF·cm-2)、x= 2 (1.96 mF·cm-2)和x= 2.5 (1.08 mF·cm-2)四组催化剂。显然,x= 1.5的催化剂为OER反应提供了更大的电活性表面,对其优异的OER活性做出了重要贡献。同时采用EIS研究了催化剂在电化学反应过程中的电子转移电阻。如图9(d)电化学阻抗(Electrochemical Impedance Spectroscopy,EIS)与表1拟合电荷转移电阻结果所示,与x= 0, 1, 2,2.5四组催化剂相比,x= 1.5对应的催化剂上电荷转移电阻明显最小。因此,当x=1.5时得到最高OER活性,在x由1到2.5增加过程中,OER活性先增加后降低,呈现火山型变化趋势。
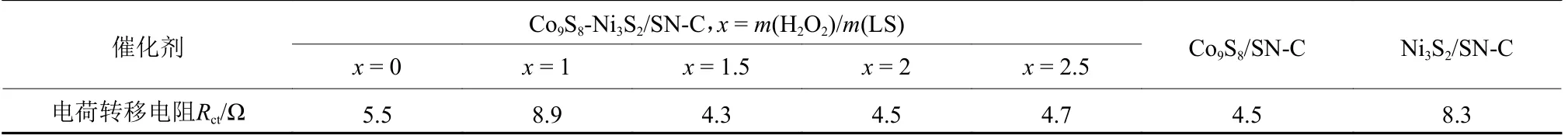
表1 Co9S8-Ni3S2/SN-C、Co9S8/SN-C与Ni3S2/SN-C的电荷转移电阻汇总表Table 1 The charge transfer resistance of Co9S8-Ni3S2/SN-C, Co9S8/SN-C and Ni3S2/SN-C
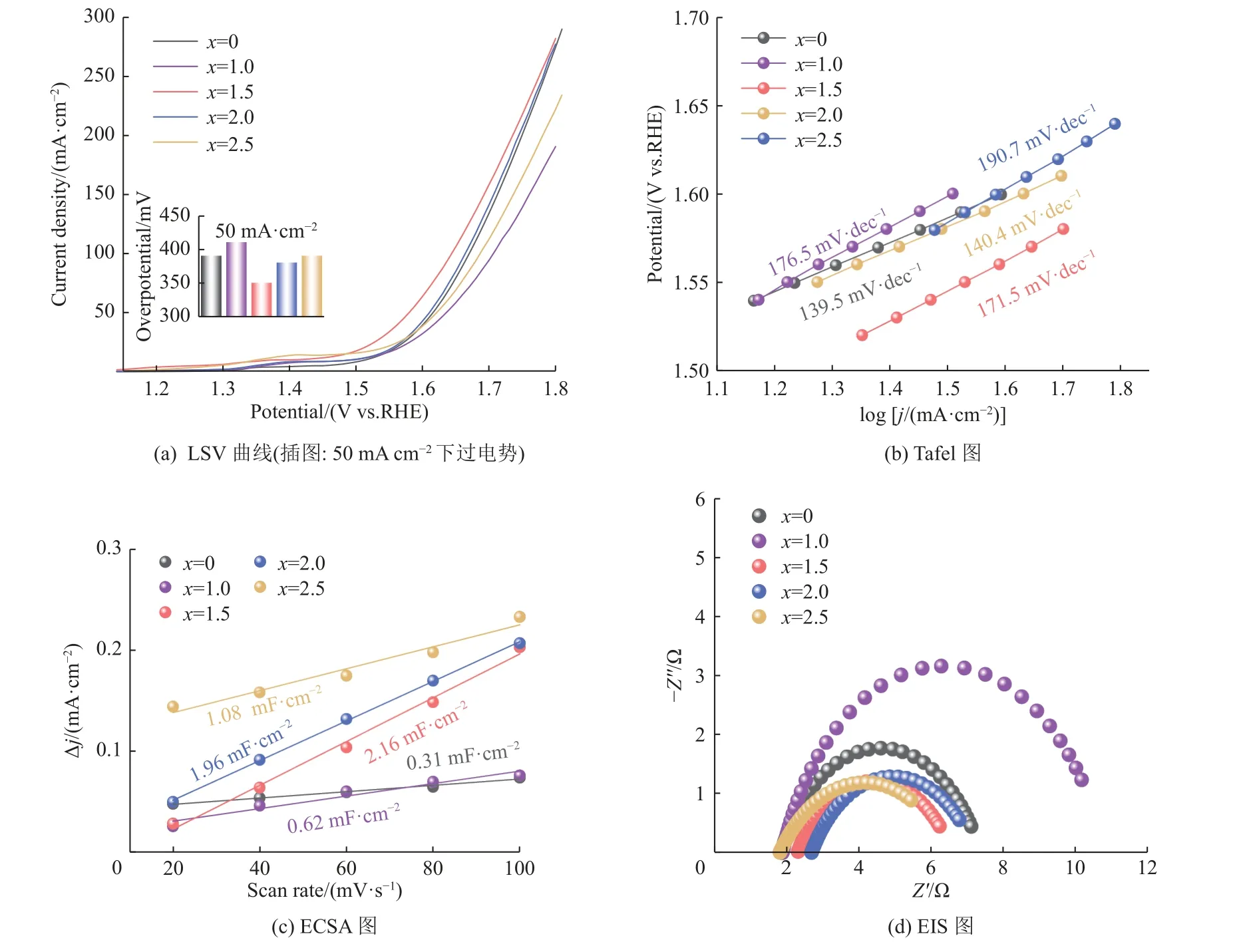
图9 LS的不同氧化氨解程度催化剂的电化学活性结果Fig.9 The electrochemical performance of catalysts prepared with different degrees of oxidative amination of LS
为了进一步验证双金属催化剂Co9S8-Ni3S2/SNC的优越性,分别构建了单金属Co9S8/SN-C和Ni3S2/SN-C催化剂并对其活性进行了对比。如图10所示,商业催化剂R u/C 的过电位(4 2 0 m V,5 0 mA·cm-2)明显高于Co9S8-Ni3S2/SN-C,证实了Co9S8-Ni3S2/SN-C催化剂的优越性。相比于Co9S8-Ni3S2/SNC催化剂,Co9S8/SN-C和Ni3S2/SN-C有着更高的过电位(380 mV,400 mV)、更高的电荷转移电阻Rct(4.5 Ω,8.3 Ω)和更小的Cdl(1.26 mF·cm-2,1.39 mF·cm-2),这表明双金属Co、Ni的强相互作用对提高催化剂的OER电催化活性有着重要作用。在50 mA·cm-2的恒定电流密度下,测试了Co9S8-Ni3S2/SN-C的电化学稳定性(见图10(d)),经过50 h的稳定性测试后电压仅增加29.1 mV,说明了催化剂良好的电催化稳定性。
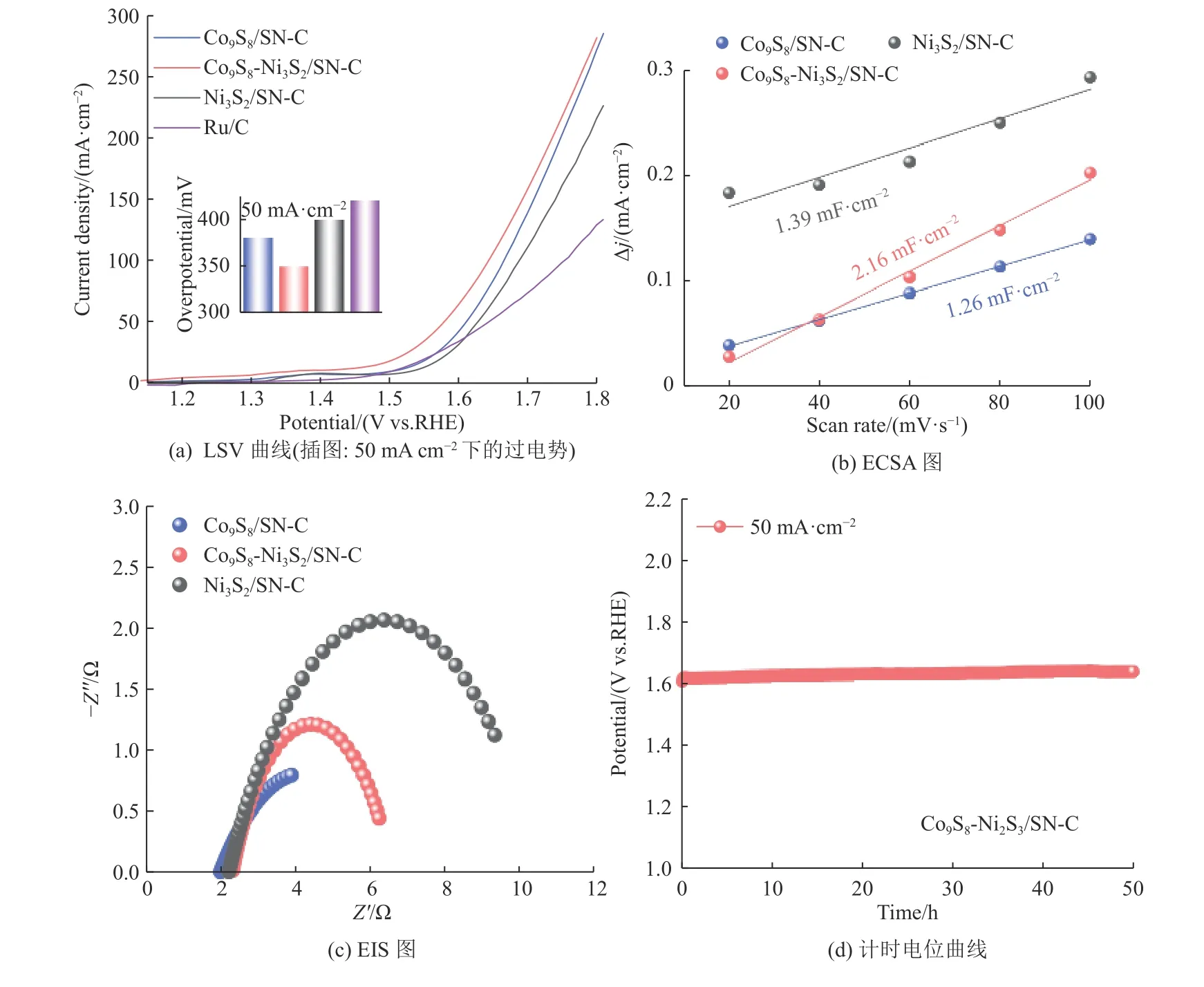
图10 Ni3S2/SN-C、Co9S8-Ni3S2/SN-C、Co9S8/SN-C和Ru/C的电化学活性与稳定性Fig.10 Electrochemical performance and stability of Ni3S2/SN-C, Co9S8-Ni3S2/SN-C, and Co9S8/SN-C
同时,通过XRD、SEM、TEM和XPS等结构表征进一步研究了经过50 h稳定性测试后的Co9S8-Ni3S2/SN-C催化剂的结构和化学稳定性。如图11(a)中SEM图所示,稳定性反应后催化剂仍保持片状结构,同时由图11(b)中HRTEM图中发现反应后的Co9S8-Ni3S2/SN-C催化剂仍具有明显的Ni3S2(202)和Co9S8(400)晶相,同时图11(c) 中EDS-Mapping结构显示Co、Ni、S、N、O和C仍均匀一致地分布。以上证明了Co9S8和Ni3S2作为主要活性结构在OER反应过程中具有较高的稳定性。不同的是,由图11(d)中的XRD图观察到,反应后Co9S8-Ni3S2/SN-C催化剂呈现出(Co, Ni) O(OH) 晶相,说明在OER反应过程中,催化剂中Co9S8和Ni3S2颗粒表面发生结构重构,对OER活性的提高具有重要作用[30]。此外,对稳定性反应后的Co9S8-Ni3S2/SN-C催化剂进行了XPS测试以分析表面化学结构变化,结果如图11(e)、(f)所示。与新鲜的Co9S8-Ni3S2/SN-C催化剂相比,反应后催化剂表面Co2+/Co3+的相对原子含量比例由2.16下降至0.58,Ni2+/Ni3+的相对含量比例由1.18下降至0.95,表明金属表面部分Ni2+和Co2+物种在反应过程中发生氧化,与XRD测试结果一致。

图11 Co9S8-Ni3S2/SN-C催化剂OER稳定性测试后的(a)SEM图;(b)HRTEM图;(c)EDS Mapping图;(d)XRD图以及(e)~(f)XPS谱图Fig.11 (a) SEM image; (b) HRTEM image; (c) EDS Mapping;(d) XRD patterns and (e)~(f) XPS spectra of Co9S8-Ni3S2/SN-C after OER stability measurement
3 总结
本文采用氧化氨解改性后的木质素磺酸钠作为S/N共掺杂碳前驱体,制备得到双金属硫化物Co9S8-Ni3S2/SN-C催化剂,具有优异的电催化OER性能。通过2D HSQC NMR、XPS、UV、FT-IR等表征确定氧化氨解改性LS与金属络合的前驱体结构,并通过TG和XRD表征分析明确了前驱体结构在热解碳化过程中的结构演变过程以及Co9S8和Ni3S2物相的形成机制。通过XRD、TEM、XPS等表征分析,证明了通过氧化氨解策略成功将配位能力较强的酰氨基引入木质素前驱体中,提高与Co、Ni离子的配位强度,改善了Co9S8-Ni3S2/SN-C催化剂上Co9S8-Ni3S2纳米颗粒的分散度,同时在Co9S8-Ni3S2二者的紧密结合与共同作用下,活性中心的能带结构得以调控;另外N的掺杂有效调控了木质素碳载体表面的电子环境,进而优化反应中间物种的吸附结构。当氧化氨解过程中m(H2O2) /m(LS) = 1.5时,Co9S8-Ni3S2/SN-C催化剂的OER活性最高,在50 mA·cm-2的电流密度下,相比商业催化剂Ru/C(420 mV)展现出更低的过电位(350 mV)。本文为二元金属硫化物电催化剂的合成提供一种绿色、低成本和通用的方法,并为实现生物质材料的高价值利用提供一个新的思路。
