UPLC-MS/MS 法同时测定补中益气丸(水丸)中6 种化合物含量
2023-11-13田小权张加稳
田小权,张加稳,王 葳
(1.昆明市药品不良反应与药物滥用监测中心,云南 昆明 650000;2.昆明市食品药品检验所,云南 昆明 650000;3.云南中医药大学,云南 昆明 650500)
补中益气丸是由李东垣精心钻研的《脾胃论》中的名方补中益气汤制成的丸剂,由炙黄芪、炙甘草、炒白术、党参、当归、陈皮、柴胡和升麻等组成,主要功效为补中益气、升阳举陷[1-3]。研究表明,补中益气丸中有效成分很多,主要包含黄酮类、皂苷类、挥发油、氨基酸及多糖类等[4],具有抗氧化、抗炎、心血管和神经保护等生理活性[5-6]。中药的药理作用是多个组分在药效上的协同作用,强调药物之间的整体作用,故中药质量评价应对处方中多个有效组分的含量进行定量分析考察。目前,2020 版《中国药典》第一部收载了补中益气丸(水丸)的质量标准,对制剂中的甘草、当归、陈皮进行薄层色谱定性分析,对黄芪甲苷单组分含量测定做了质量控制以及对炙黄芪、炙甘草、炒白术、陈皮、党参、升麻和柴胡进行显微鉴别,对其药效质量的分析评价存在局限性[7-8]。
现有文献报道多是采用高效液相色谱(highperformance liquid chromatography-tandem,HPLC)法测定补中益气丸中的活性成分含量[9],该法同时测定多种化合物时常遇到基质干扰大、易出现假阳性、分离效果差,以及耗时较长等问题。近年来,超高效液相色谱-串联质谱法(Ultra-high-performance Liquid Chromatography-tandem Mass Spectrometry,UPLC-MS/MS) 法已发展成为测定药物中多组分有机物微量分析的重要方法[10]。鉴于对补中益气丸(水丸)中多种活性成分含量同时测定的文献报道较少,本文建立UPLC-MS/MS 法同时测定黄芪甲苷、毛蕊异黄酮苷、橙皮苷、阿魏酸、甘草酸和甘草苷等6 种化合物组分含量,旨在为该制剂的药效质量分析评价提供理论指导。
1 仪器与材料
超高效液相-串联质谱联用仪(EXION-QTRAP 4500,美国AB 公司),超声提取仪(SK8210HP,上海科导公司),十万分之一电子天平(MS205DU 型,梅特勒-托利多Mettler Toledo 公司),Agilent Zorbax Eclipse XDB-C18 色谱柱(填料:十八烷基硅烷键合硅胶2.1 × 100 mm,1.8 μm,美国Agilent 公司);PES滤膜(0.22 μm,天津津腾有限公司)。黄芪甲苷(批号:110781-202118)、毛蕊异黄酮苷(111920-201606)、橙皮苷(批号:110721-202019)、阿魏酸(批号:110773-201915)、甘草酸(100551-202003)、甘草苷(批号:111610-201908)对照品均由中国食品药品检定研究院提供。补中益气丸(水丸)由同药集团大同制药有限公司制作,批号:171002。甲酸(分析纯,国药集团化学试剂有限公司),乙腈(色谱纯,默克股份两合公司)。
2 方法与结果
2.1 仪器条件
2.1.1 液相色谱条件 流速:0.35 mL/min;柱温:40 ℃;进样量:10 μL;流动相A 为5 mmol/L 乙酸铵水溶液,流动相B 为甲醇(含0.1%甲酸)。洗脱梯度见表1。
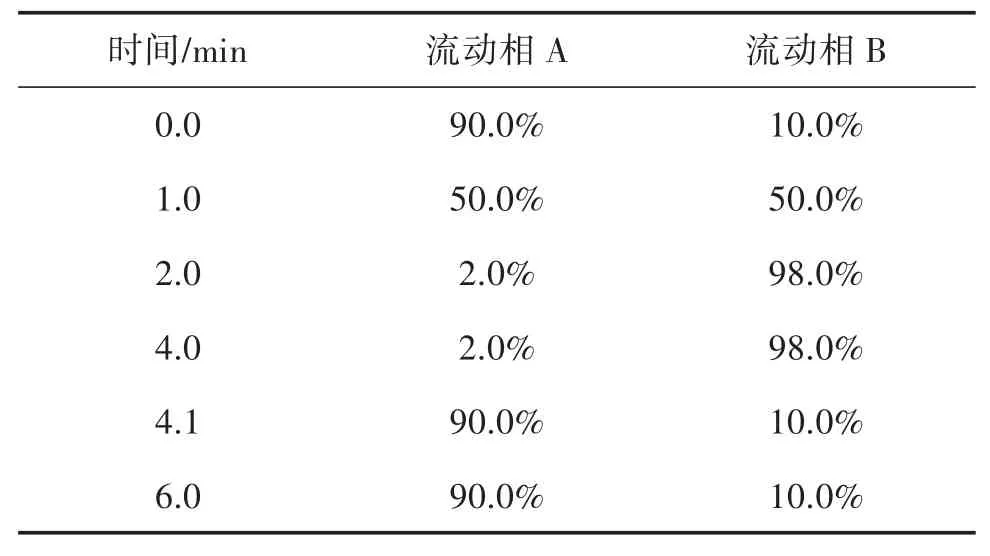
表1 洗脱梯度
2.1.2 质谱条件 正、负离子扫描;MRM 多反应监测;气帘气、离子源雾化气、离子源辅助加热气分别设定为38 psi、55 psi 及55 psi,喷雾电压为5 500 V,加热器温度为550℃;优化后的监测离子对(m/z)、去簇电压(DP)、碰撞电压(CE)等结果参数见表2。
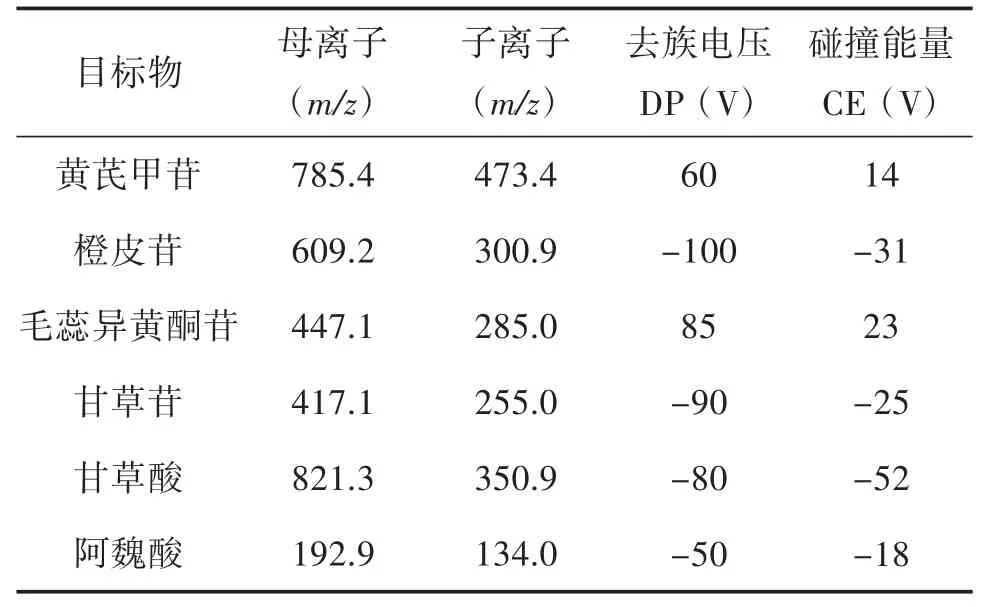
表2 6 种化合物的MRM 质谱参数[11]
2.2 溶液制备
2.2.1 对照品溶液 精密称取各标准品约10 mg,用甲醇溶解并定容,制成质量浓度均为1.0 mg/mL 混合标准储备液;分别精密量取适量储备液混合置容量瓶中,以70%甲醇溶液稀释,制得标准品系列溶液,避光4 ℃保存备用。
2.2.2 供试品溶液 取本品适量,研细至粉末状,精密称取0.5 g,将其置于具塞锥形瓶中,精密加入70%甲醇25 mL,紧密封塞,摇匀,称定重量,将超声仪温度调至30 ℃,超声45 min,待锥形瓶冷却至室温后,补足重量,摇匀,过滤,精密量取25 mL,置100 mL 容量瓶中,70%甲醇定容至刻度线,即得供试品溶液。
2.3 线性关系考察 所测成分的定量下限(LLOQ)要求信噪比S/N>10,6 次进样,计算平均峰面积精密度RSD≤10%。根据样品含量预实验测定,选择6 个浓度的混合对照品溶液重复进样(n=2),以目标物定量离子对的平均峰面积计算进样质量浓度(ng/mL)得线性回归方程。6 种化合物的标准混合液和样品液的MRM 离子图分别见图1 和图2,回归方程线性范围、相关系数(r2)及LLOQ 见表3。结果显示,在相应浓度范围内各化合物线性关系良好(r2为0.994~0.999)。

图1 6 种化合物标准液的MRM 质谱图
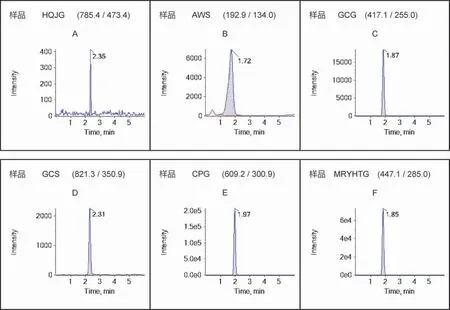
图2 样品液中6 种化合物的MRM 质谱图
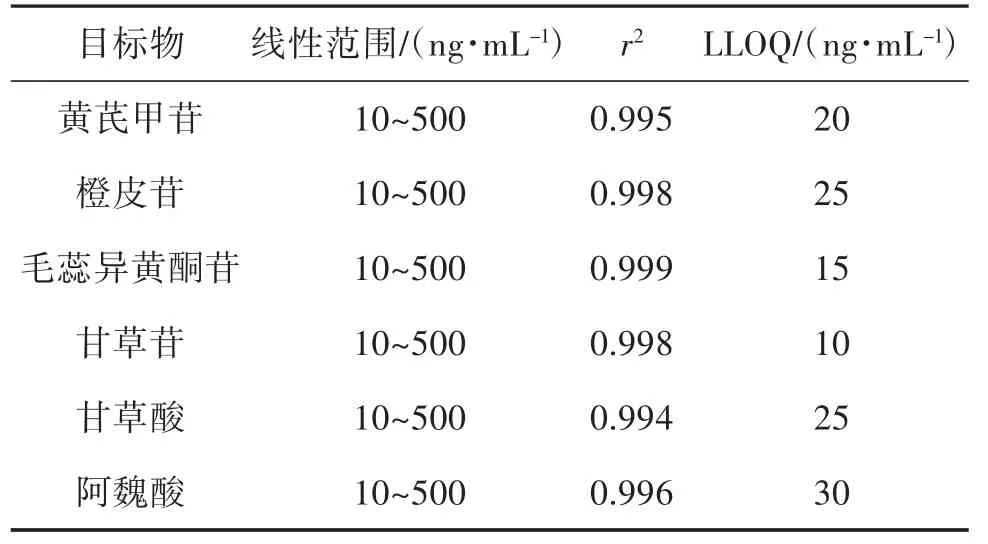
表3 6 种化合物的线性范围、r2 和定量下限
2.4 精密度试验 取质量浓度为50 ng/mL(中浓度)的混合对照品溶液,连续进样6 次,测定黄芪甲苷、毛蕊异黄酮苷、橙皮苷、阿魏酸、甘草酸和甘草苷6 种化合物的平均峰面积,依次为2 189、5 616、3 471、3 187、3 288、7 364,计算得6 种化合物平均峰面积的RSD 分别为1.7%、2.3%、4.1%、5.6%、4.7%、3.8%。
2.5 重复性试验 按照“2.2.2”项下方法制备6 份供试品溶液,分别测定得到样品中黄芪甲苷、毛蕊异黄酮苷、橙皮苷、阿魏酸、甘草酸和甘草苷6 种化合物含量的RSD 分别为5.3%、7.5%、6.7%、8.3%、6.0%、4.2%,结果呈现良好的方法重复性。
2.6 稳定性试验 取浓度为100 ng/mL 的混合标准品溶液,依次进样测定存放第0、2、4、8、12、24、48 h的溶液中黄芪甲苷、毛蕊异黄酮苷、橙皮苷、阿魏酸、甘草酸和甘草苷峰面积的RSD,结果分别为1.2%、2.5%、1.8%、3.2%、1.5%、0.9%,表明总体稳定性良好,但阿魏酸稳定性略偏低,可能是试样前处理时被氧化。
2.7 加样回收试验 称取已测定的补中益气丸约0.5 g,精密加入低、中、高3 种质量浓度的储备混合液适量,每组平行6 份,按照“2.2.2”项下方法制备,依次测定各组分含量,计算回收率。结果显示黄芪甲苷、毛蕊异黄酮苷、橙皮苷、阿魏酸、甘草酸和甘草苷的平均回收率分别为98.4%、101.7%、101.4%、86.7%、103.5%、93.7%,对应的RSD 在3.8%~7.6%之间。
2.8 样品含量测定 称取补中益气丸(水丸)供试品约0.5 g,按“2.2.2”项方法制备6 份供试液,分别分析供试品中黄芪甲苷、毛蕊异黄酮苷、橙皮苷、阿魏酸、甘草酸和甘草苷的平均含量。结果见表4。
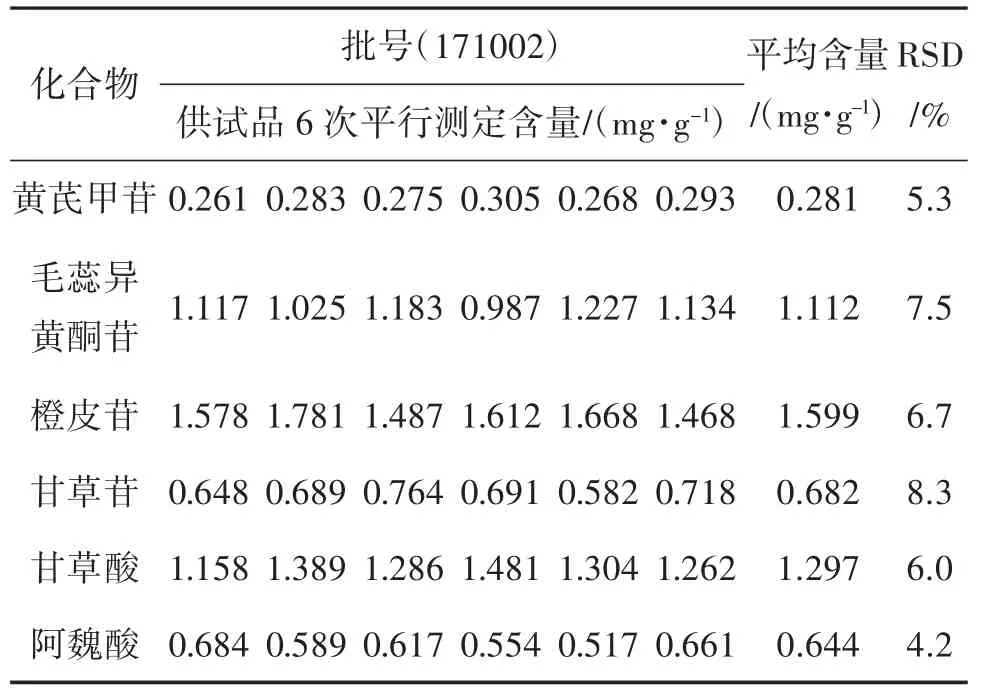
表4 供试品中6 种组分的含量测定结果
3 讨论
3.1 供试品溶液制备 根据补中益气丸(水丸)中6 种化合物组分的提取效率,对提取方法作了优化研究:比较了30%甲醇、50%甲醇、70%甲醇、纯甲醇对供试液中6 种化合物提取效率的高低,结果表明70%甲醇更高,且峰形更好,原因可能是各化合物极性与该比例提取溶剂相近,流动相洗脱效率更高;比较了对供试液超声10 min、20 min、30 min、45 min、60 min,结果表明超声45 min 和60 min 提取效率明显更高,但两者无明显差异,故选择超声时间为45 min。
3.2 色谱条件优化 依据化合物性质和色谱条件,研究对比了乙腈-水、甲醇-水、甲醇-乙酸铵-水、甲醇-甲酸-水、含0.1%甲酸的甲醇-乙酸铵-水组成的流动相体系的洗脱效果。结果表明,经优化后含0.1%甲酸的甲醇-乙酸铵-水混合体系对6 种化合物的洗脱效果更好,峰形对称、分离度高。对超高效液相色谱柱Agilent Zorbax Eclipse XDB-C18、MAbPac SCX-10 和ProPac Elite WCX 进行分析,比较色谱峰形和分离度,结果显示经Agilent Zorbax Eclipse XDBC18 洗脱峰形更好,且出峰时间最短。
4 结论
本文建立了同时测定补中益气丸(水丸)中6 种生物活性成分的UPLC-MS/MS 定量分析方法,其操作简单、快速,6 种化合物的平均回收率均在86.7%~103.5%,相对标准偏差均小于10%,表明该方法准确度高、重复性好。本实验为补中益气丸(水丸)中多组分定量检测的质量分析评价提供了参考。
