气相色谱-质谱法测定西吡氯铵含片中的氯代烷烃类杂质
2023-11-04刘延凤卢红华孙建合伊磊张贵民
刘延凤,卢红华,孙建合,伊磊,张贵民
(鲁南制药集团股份有限公司,国家手性制药工程技术研究中心,山东 临沂 273400)
西吡氯铵是一种阳离子季铵化合物,用于治疗单纯性牙龈炎,作为首选机械性去除局部病因的方法,对牙齿无着色,对口腔粘膜无刺激,能减少牙菌斑的形成[1-2]。西吡氯铵以吡啶、氯代十六烷为起始物料,一步反应合成。氯代十六烷为具有警示结构的卤代烷烃类化合物[3],还可能会引入相近链长的氯代十四烷、氯代十五烷、氯代十七烷与氯代十八烷[4]。西吡氯铵含片中氯代烷烃类杂质的研究尚未有文献报道,其中氯代十六烷既是工艺杂质又是可能产生的降解产物,需要重点关注。根据人用药品注册技术要求国际协调会(International Conference on Harmonization,ICH)M7[5]指南规定,单官能团烷基氯化物的TD50在36~108 mg/(kg·d),按更为严格的36 mg/(kg·d),根据西吡氯铵含片的最大日服用量8 mg·d-1,用线性外推法计算,以上氯代烷烃类杂质的限度均不得过标示量的0.45%,严控为0.1%。本研究采用气相色谱-质谱联用法(GC-MS)检测西吡氯铵含片中氯代烷烃类杂质的含量,用萃取法处理样品,使水溶性好的西吡氯铵和水溶性差的待测成分分别溶于水相和有机相,避免了西吡氯铵进入色谱仪遇热分解以致待测成分测得量偏离实际值,同时,采用硝酸银沉降水相中的氯离子,进一步消除了西吡氯铵中十六烷基降解生成氯代十六烷的反应条件。
1 仪器与材料
1.1 仪器
7890B-7000D型气相色谱质谱仪(美国安捷伦公司);MS205DU型电子分析天平(瑞士梅特勒公司)。
1.2 材料
氯代十四烷对照品(纯度:98.3%,阿拉丁试剂有限公司);氯代十五烷对照品(纯度:99.4%,山东新时代药业有限公司);氯代十六烷对照品(纯度:98.0%,上海毕得医药科技股份有限公司);氯代十七烷对照品(纯度:99.3%,山东新时代药业有限公司);氯代十八烷对照品(纯度:98.3%,上海麦克林生化科技股份有限公司);西吡氯铵含片(山东新时代药业有限公司,批号220501、220502、220503,含量>98.0%);正己烷(德国默克公司,色谱纯);硝酸银(国药集团股份有限公司,分析纯)。
2 方法与结果
2.1 色谱及质谱条件
色谱柱HP-5MS毛细管柱(30 m×0.25 mm,0.25 μm),以5%苯基-95%甲基聚硅氧烷为固定液;初始柱温为60 ℃,保持1 min后,以30 ℃·min-1的速率升温至300 ℃,保持3 min;进样口温度350 ℃;载气为氦气;柱流速1.0 mL·min-1;分流比10∶1;进样量1 μL。电子轰击离子化(EI)模式电离,多反应监测模式(MRM)定量分析;离子源电压-70 eV;离子源温度230 ℃;接口温度300 ℃;监测离子对m/z 91→55。
2.2 溶液配制
2.2.1 空白溶剂
取正己烷作为空白溶剂。
2.2.2 对照品溶液
分别取各杂质对照品约13 mg,精密称定,置量瓶中,用正己烷定容稀释制成每1 mL中各约含1.3 μg的混合溶液,作为对照品贮备液。精密量取对照品贮备液0.1 mL,置10 mL量瓶中,用正己烷稀释至刻度,摇匀,即得。
2.2.3 供试品溶液
取西吡氯铵含片研细,取细粉约25 mg(相当于西吡氯铵0.067 mg),精密称定,置具塞玻璃试管中,精密加入5 mL硝酸银溶液(取硝酸银约200 mg,置100 mL量瓶中,加水溶解并稀释至刻度,摇匀),超声5 min,精密加入5 mL正己烷萃取,取上清液,即得。
2.3 专属性
取“2.2”项下空白溶剂和对照品溶液,分别依法测定,记录色谱图。结果显示,空白溶剂对待测成分无干扰;对照品溶液中,氯代十四烷的保留时间为8.098 min、氯代十五烷的保留时间为8.489 min、氯代十六烷的保留时间为8.861 min、氯代十七烷的保留时间为9.217 min、氯代十八烷的保留时间为9.574 min,各组分间均能较好分离。
2.4 精密度、重复性试验
精密量取“2.2”项下对照品贮备液0.1 mL置10 mL量瓶中,用正己烷定量稀释至刻度,摇匀,重复测定6次,计算氯代十四烷、氯代十五烷、氯代十六烷、氯代十七烷与氯代十八烷峰面积的RSD分别为1.23%,1.08%,1.19%,1.59%与2.01%。结果表明,本法精密度良好。
取西吡氯铵含片(批号220501)细粉约25 mg,精密称定,置具塞玻璃离心管中,精密加入5 mL硝酸银溶液,加入对照品贮备液100 μL,超声5 min,精密加入5 mL正己烷萃取,取上清液,平行配制6份,依法测定。氯代十四烷、氯代十五烷、氯代十六烷、氯代十七烷与氯代十八烷测得含量的RSD分别为0.097%,0.10%,0.13%,0.06%与0.22%。结果表明,本法重复性良好。
2.5 线性与范围
精密量取对照品贮备液1 mL置10 mL量瓶中,用正己烷定量稀释至刻度,摇匀,作为线性贮备液。精密量取线性贮备液0.2,0.4,0.6,0.8,1.0,1.2 mL分别置10 mL量瓶中,用正己烷稀释至刻度,摇匀,作为线性系列标准溶液。分别依法测定,记录色谱图。以各组分的质量浓度c(ng·mL-1)为横坐标,峰面积A为纵坐标进行线性回归,回归方程为氯代十四烷:A=22.296c+1.623,r=0.999 9;氯代十五烷:A=16.256c+0.393,r=0.999 8;氯代十六烷:A=10.528c+3.443,r=0.999 6;氯代十七烷:A=6.967c-0.312,r=0.999 8;氯代十八烷:A=5.337c-2.148,r=0.999 7。结果表明,待测成分在质量浓度2.6~26 ng·mL-1内,线性关系良好。
2.6 回收率
取西吡氯铵含片(批号220501)细粉约25 mg,精密称定,置具塞玻璃离心管中,精密加入5 mL硝酸银溶液,分别加入对照品贮备液80,100,120 μL,超声5 min,分别精密加入5 mL正己烷萃取,取上清液,作为80%~120%回收率试验溶液,每个浓度制备三份。分别依法测定,记录各成分的峰面积。结果显示,各成分的平均回收率(n=9)与RSD分别为氯代十四烷:101.28%,2.89%;氯代十五烷:101.96%,3.39%;氯代十六烷:102.02%,2.1%;氯代十七烷:101.30%,3.1%;氯代十八烷:102.55%,3.8%;方法准确度高。
2.7 定量限与检测限
取各成分对照品溶液逐级稀释,分别以信噪比S/N为10和3时的溶液作为定量限溶液与检测限溶液。定量限溶液重复测定6次,氯代十四烷、氯代十五烷、氯代十六烷、氯代十七烷和氯代十八烷峰面积的RSD(n=6)依次为:3.1%,3.2%,3.5%,3.1%和3.0%;定量限质量浓度依次为0.82,1.27,1.35,3.54和4.39 ng·mL-1;检测限质量浓度依次为0.41,0.64,0.68,1.77和2.20 ng·mL-1;表明本法灵敏度高。
2.8 稳定性
取“2.2”项下对照品溶液与供试品溶液,分别于室温放置0,2,4,6,8,10,12 h时,按“2.1”项下条件进样测定。结果显示,12 h内待测成分峰面积的RSD均小于10.0%,保留时间的RSD均小于2.0%,表明对照品溶液与供试品溶液在12 h内稳定性良好。
2.9 耐用性
先后改变初始柱温(58,62 ℃)、升温速率(28,32 ℃·min-1)、柱流速(0.9,1.1 mL·min-1),取“2.2”项下空白溶剂、对照品溶液与100%回收率溶液,分别依法测定,考察样品中待测成分检出情况。结果表明,不同耐用性条件下,待测成分检出结果与标准条件无明显差异,耐用性好。
2.10 样品测定
取三批样品,按“2.2”项下方法配制对照品溶液与供试品溶液,按“2.1”项下条件进样测定,按外标法以峰面积计算。图1结果显示三批样品中待测成分均未检出。
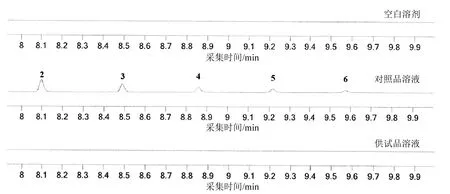
图1 典型色谱图
3 结论
西吡氯铵含片中载药量较低,受辅料影响,难以配制得到含较高浓度西吡氯铵的样品,采用液相色谱法、气相色谱法灵敏度均不能达到检测要求。采用气质联用法,随进样口温度升高,氯代十六烷的检出含量逐渐增大,表明样品在较高的汽化温度下降解;而进样口温度太低,沸点较高的待测成分不能完全汽化,重复性差。本研究中,创新性地采用萃取法配制样品,利用西吡氯铵与氯代烷烃类杂质的溶解性差异,使两者分置两相溶液中,取含有待测组分的有机相作为供试品溶液,既避免了样品降解对测定结果的干扰,又可达到准确检验待测组分的目的。于水相中加入硝酸银,使氯离子沉降,消除了样品降解的条件,同时,盐析效应提高了萃取效率。经试验,对照品溶液萃取前后待测成分的浓度无明显差异,回收率接近100%,表明待测成分可被充分萃取进入有机相。采用离子监测模式对本法的线性与范围、精密度、回收率、定量限与检测限、溶液稳定性进行了验证,结果显示,本法的准确性好、灵敏度高、溶液稳定性好,可用于西吡氯铵含片中氯代烷烃类杂质的检测。
