成人MOG抗体病和抗NMDAR脑炎叠加综合征1例并文献复习
2023-11-02高曲文黎振声齐自娟邓彩云项薇邓兵梅
高曲文,黎振声,齐自娟,邓彩云,项薇,邓兵梅
抗N-甲基-D-天冬氨酸受体(Anti-N-methyl-D-aspartate receptor,NMDAR)脑炎是脑脊液中存在抗NMDAR RN1(GluN1)亚基抗体的自身免疫性脑炎,其主要临床症状包括精神异常、癫痫发作、记忆下降等[1]。髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)仅表达于中枢神经系统少突胶质细胞质膜上,位于髓鞘最表面。MOG抗体病是一种独立的急性中枢神经系统脱髓鞘疾病,可伴有双侧视神经炎、脑干脑炎、长节段性脊髓炎和类似急性播散性脑脊髓炎的临床表现[2]。近年来,MOG抗体病与抗NMDAR脑炎叠加综合征颇受关注,但国内报导多为儿童病例,成人相对少见。另外,2 种抗体并存的具体机制仍不清楚。在此,我们报道1例成人病例并检索相关文献,总结成人MOG抗体病与抗NMDAR脑炎叠加综合征的临床特点、影像学表现及治疗情况,并对2种抗体并存的可能机制进行探讨。
1 资料与方法
1.1 病例资料
患者,男,22岁,既往身体健康,因“发作性右侧面部麻木、右上肢抽搐18天”于2018年2月23日入院。患者于2月5日午餐后无明显诱因出现右侧面部麻木,嘴角歪斜,约1 min 后出现右上肢抽动,双下肢无力,随后摔倒,持续约2~3 min后症状消失,无意识丧失。病前1 周有鼻塞及咽痛史。2 月6 日就诊于我院门诊,脑MRI示左颞内侧FLAIR高信号灶伴周围低信号及同侧颞角消失,怀疑节细胞胶质瘤(图1A)。上述发作2~3 次/d,2 周后出现1 次继发全身性强直阵挛发作发作(图2)。入院查体:神志清楚,言语欠流利,右上肢近端肌力Ⅳ级,远端肌力Ⅳ-,右侧面部及右侧上肢第1~4指及虎口、大鱼际痛温觉、触觉较对侧减弱。入院后查心电图、胸片无异常,血氨、三大常规、全生化、血电解质、免疫5项、血管炎5项、ENA谱、凝血四项、输血前八项大致正常。为判断肿瘤性质行脑PET/CT 检查,结果示左颞低代谢,左额、中央区高代谢(图1D、E)。脑脊液检查,白细胞15 个/μL,生化正常,IgG 指数0.62。视频脑电图示间期C3、P3导联持续性慢波活动,伴有小尖棘波。MRI复查示左侧额、中央区脑沟消失,FLAIR高信号(图1B)。自身免疫性脑炎6项检测示血清及脑脊液抗NMDAR抗体IgG均阳性(图2)。肺部CT、腹部B超、睾丸、附睾B超、肿瘤蛋白芯片未见异常。诊断为抗NMDAR抗体脑炎,予以丙球(0.4 g/kg/d×5 d),并联合激素冲击治疗(甲强龙1000 mg,1 次/d,3 d 减半),后继口服激素逐渐减量(图2)。经治疗后临床症状明显好转,无发作。3个半月后,强的松减至10 mg/d,头颅MRI 复查示左侧颞叶内侧病变较前明显变小,左侧前扣带回新发病灶(图1C),PET/CT相应部位未见高代谢(图1F)。

图1 患者脑MRI及PET/CT表现

图2 患者临床经过
2018年7月,停用口服激素10 d后,患者感头昏、视物不清(左眼明显)再次入院(图2)。复查MRI 示桥脑被盖、双侧视束(左侧明显)、胼胝体前部、左侧顶叶白质FLAIR 高信号(图1G-J)。复查腰穿及相关抗体:脑脊液生化正常,白细胞45个/μL,IgG 指数1.1,脑脊液抗NMDAR 抗体、抗MOG 抗体及血抗MOG 抗体均阳性(图2)。诊断考虑MOG 抗体病合并抗NMDAR 脑炎,再予激素冲击治疗,后继激素逐渐减量,并予以吗替麦考酚酯口服(750 mg,2 次/d)。2 周后头昏、视物不清均较前明显好转。
2018年12月初,强的松减至10 mg/d,患者出现喉部疼痛,后感头昏、头痛,复查头颅MRI发现左侧桥臂、左侧视束区新发病灶(图1K、L)。再次予丙球冲击治疗,吗替麦考酚酯加量至1000 mg,2次/d,出院时头痛情况较前有所好转。
2019年1月,患者生活不规律,经常熬夜饮酒,自行将吗替麦考酚酯减至500 mg,2 次/d。2019 年4 月,出现头昏、视物不清、行走不稳(图2)。查体示:左侧眼裂较右侧小,双眼侧视时可出现水平眼震,向左侧视时明显,闭目难立征阳性。脑MRI的FLAIR 像示右侧桥脑、桥臂、丘脑,左侧枕叶新发高信号灶(图1M、N)。考虑自身免疫性脑炎复发,再次予以丙球冲击并将强的松加量至60 mg/d。经调整治疗后患者临床症状明显缓解。
2019 年12 月,患者自行停用所有药物包括口服强的松、吗替麦考酚酯及抗癫痫药物。2020年1月,出现头昏、行走不稳,查体示:右下肢深感觉减退,Romberg 征(±)。复查脑MRI,FLAIR 像示延髓右侧、右颞深部、左侧半卵圆中心新发高信号灶(图1O-Q)。脑脊液复查示白细胞10 个/μL,IgG 指数0.47。复查血及脑脊液抗NMDAR 和抗MOG 抗体均为阳性(图2)。考虑MOG 抗体合并抗NMDAR 脑炎复发,再次予以丙球及激素静脉滴注后症状改善。出院时仍有视物模糊,无头昏、行走不稳,嘱规律生活、按时服药及定期复诊。
1.2 方法
在查询框中选“题名/摘要(Title/Abstract)”,以“MOG”、“NMDAR”为关键词,逻辑关系选“和(AND)”在CNKI、Sinomed数据库及PubMed(建库至2021年4 月21日)检索中英文文献,阅读所检索到的文献,并结合文中提及的参考文献检索相关参考文献。
2 结果
共筛选出年龄≥18岁的MOG抗体病与抗NMDAR脑炎叠加综合征病例15 例(见表1)。抗NMDAR 脑炎诊断标准符合2016年自身免疫性脑炎临床诊断国际专家共识[3],MOG抗体病诊断标准参考2018年美国梅奥诊所专家组提出的MOG抗体病诊断标准[4]。MOG 抗体病与抗NMDAR 脑炎叠加综合征的诊断标准为[5]:①临床符合抗NMDAR脑炎和(或)MOG抗体病诊断标准;②至少1 次急性发作期脑脊液抗NMDAR 抗体及血或脑脊液MOG抗体同时阳性。排除标准:①临床资料不全;②年齡<18 岁;③随访时间<3 个月;④其他系统性自身免疫性疾病。
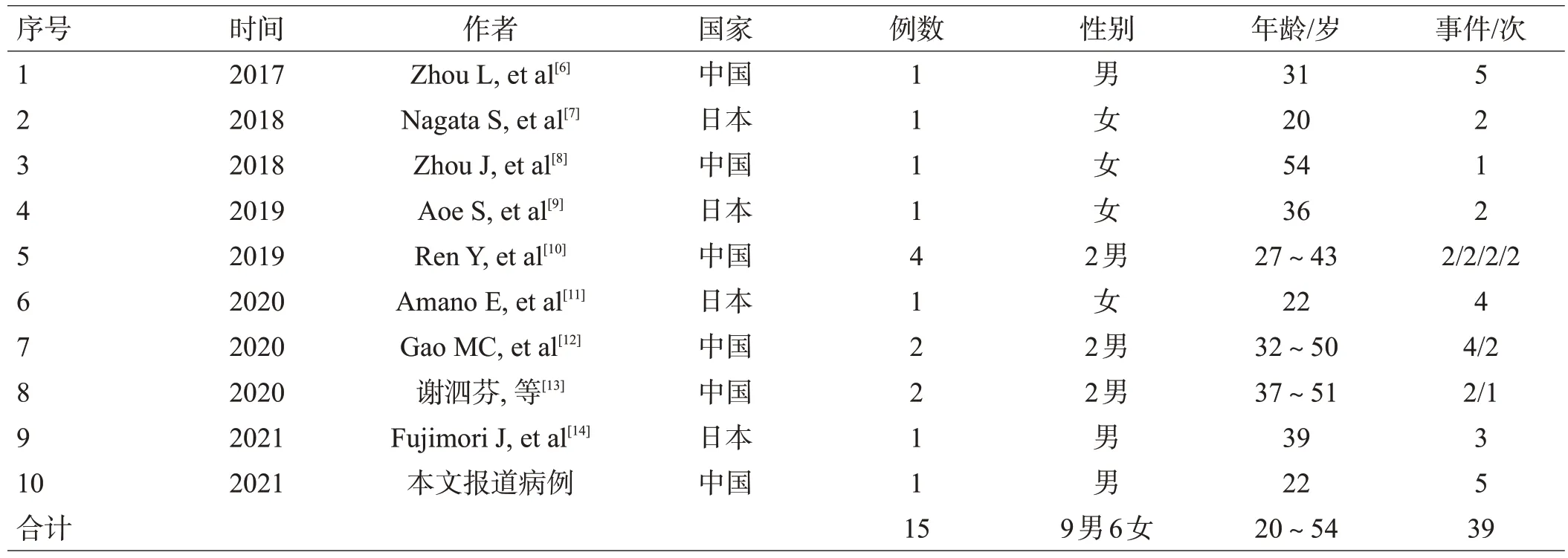
表1 成人MOG抗体病和抗NMDAR脑炎叠加综合征病例汇总
15例患者中,男性9例,女性6例,主要来自中国和日本,平均始发年龄34.7(20~54)岁,呈复发缓解病程13例(86.7%)。15例患者共有39次发作事件,平均复发间隔时间(14.4±24.4)个月。
首发症状以头痛最多见,其次为抽搐、发热5(表2)。15例患者共39 次急性发作,临床症状以头痛最常见,其次为视力下降、精神行为异常(表2)。

表2 成人MOG抗体病和抗NMDAR脑炎叠加综合征病例临床症状汇总
临床分型:39 次急性发作中MOG 抗体病1 例次,抗NMDAR 脑炎4 例次,未定型10 例次,重叠型24 例次。24 例次重叠型急性发作临床症状排前3的依次为视力下降、头痛、精神行为异常(表2)。
辅助检查:①急性期血及脑脊液抗体检测:15 例患者中,MOG 抗体脑脊液检测20 例次,阳性16 例次(80%);MOG 抗体血清学检测27 例次,阳性21 例次(77.8%)。②影像学检查:共收集急性发作期头颅MRI 33 次,异常32 例次,其中皮质受累32例次、脑干12例次、放射冠8例次、视神经或视束7例次、小脑5例次。
治疗及愈后:15例患者23次发作急性期均予大剂量甲泼尼龙治疗,后改为口服泼尼松序贯治疗。14 例次给予静滴人免疫球蛋白治疗。2 例次给予血浆置换。15 例患者对一线免疫治疗均敏感,5 例患者在激素减量过程中出现复发,8 例患者加用二线免疫抑制剂治疗,主要包括硫唑嘌呤(5例)、吗替麦考酚酯(4 例)。1 例患者手术切除卵巢畸胎瘤。15 例患者中有3 例依丛性较差,1例半年随访时仍有双眼视物模糊及被害妄想,改良Rankin 量表(modified Rankin Scale,mRS)评分3 分;1 例右眼视力下降,MRS 1 分;本文病例1 年后随访时仍遗留双眼视力下降,MRS 1分。其余13例MRS评分0~1分10例,2分2例。
3 讨论
本文报告1 例成人MOG 抗体病和抗NMDAR 脑炎叠加综合征。文献复习共收集到15 例成人病例,共39 次急性发作事件。始发症状以头痛、抽搐、发热多见,急性发作症状以头痛、视力下降、精神行为异常最多见。临床分型以叠加型为主,最常见的症状依次为视力下降、头痛。而儿童MOG 抗体病和抗NMDAR 脑炎叠加综合征无论是始发症状、急性发作症状及叠加型症状均以抽搐、嗜睡最为多见[5]。成人与儿童MOG抗体病和抗NMDAR脑炎叠加综合征临床表现的差异的原因可能与儿童大脑成熟度没有成人高及免疫系统尚未完成成熟有关。
在15 例患者中,MOG 抗体在脑脊液中的阳性率达80%,提示血MOG 抗体阴性不能排除合并MOG 抗体病的可能,当怀疑合并MOG 抗体病时最好能同时行脑脊液和血MOG抗体检测[14]。15 例患者均有MRI 异常,约60%患者受累部位在2 个以上,以皮质、脑干、放射冠、视神经最为多见,可能与NMDAR主要分于皮质及MOG位于中枢神经髓鞘有关。所有患者对激素治疗敏感,约1/3患者在激素减量过程中出现复发,大部分病例合并使用2 种以上免疫治疗措施,提示该叠加综合征可能需要更强或更长时间的免疫治疗[2]。经治疗后约80%的患者预后较好,且与患者对治疗依丛性有一定关系,中断或不配合免疫治疗的患者预后较差,谢泗芬等[13]亦报导了类似现象。
MOG抗体和NMDAR抗体共存的机制目前尚未明确,可能有以下几个方面的原因:①MOG和NMDAR共存于少突胶质细胞表面,在针对少突胶质细胞的自身免疫期间,免疫细胞可能会攻击到同一位置的MOG和NMDAR自身抗原,从而在脑脊液和血清中产生相应的抗体;②在抗NMDAR脑炎患者中,MOG抗体的阳性率约为3.3%~17%[15-17],远高于普通人群中MOG抗体的阳性率(1.9/100,000)[18];另外,MOG 抗体病患者中抗NMDAR的阳性率约为8.8%[19],亦远高于普通人群(0.6/100,000)[18]。这些提示抗NMDAR脑炎或MOG抗体症可能促进另一疾病的发生。Titulaer等[15]认为抗NMDAR脑炎与MOG抗体症可能存在共同的发病机制,即某些诱因导致体液免疫紊乱产生同时针对MOG 和NMDAR 的自身抗体。③免疫重构的影响。在减少或停止免疫治疗时,免疫系统从免疫抑制状态中恢复并重建,可能导致免疫细胞攻击自身抗原而引起中枢神经系统炎性反应[20]。
我们的病例主要来自中国和日本,可能主要受检索及排除标准所致,亦可能与不同国籍作者写作偏好有关,由于病例数量少不足以说明该病与人群种族因素有关,另外本文结论可能存在一定偏倚。
综上所述,成人MOG 抗体病和抗NMDAR 脑炎叠加综合征并不少见,以男性多见,大多呈复发缓解病程,发作期症状以头痛、视力下降、精神行为异常多见,头颅MRI 阳性率高,主要累及皮质、脑干、放射冠、视神经,所有患者对一线免疫治疗敏感,大多需要2种以上的免疫治疗方法且易反复,可能需要更强或更长时间的免疫治疗。建议临床诊断为抗NMDA 受体脑炎或MOG抗体病患者行血清及脑脊液检查时同时进行自身免疫性脑炎相关抗体及中枢神经系统脱髓鞘疾病相关抗体的检测,以制定最佳的治疗方案以减少复发。
