燕麦红叶病介体蚜虫获毒前后差异表达基因分析
2023-10-28魏淑花马建华朱猛蒙
王 颖,魏淑花,张 蓉,马建华,朱猛蒙
(宁夏农林科学院植物保护研究所,银川 750002)
燕麦(Avena sativaL.)是一年生粮饲兼用作物,具有耐寒、抗旱、耐贫瘠、耐盐碱、适应性强的特性,燕麦叶片、秸秆多汁柔嫩,适口性好,秸秆中粗蛋白、粗脂肪及无氮抽出物含量高,难消化纤维含量低,是宁夏回族自治区(以下简称宁夏)奶业高质量发展的重要饲草支撑,对稳定中国畜牧业发展、平衡农业种植结构和生态环境保护具有重要作用[1-3]。
随着燕麦的大面积种植及燕麦产业的不断发展,病虫害的发生愈发严重[4]。其中,燕麦红叶病是由蚜虫传播大麦黄矮病毒(Barley yellow dwarf virus,BYDV)引起的病毒病。该病不仅会引起燕麦茎叶发红发紫、碳氮代谢紊乱、根际硝酸盐累积、植株韧皮部组织坏死等,而且会造成植株矮化、分蘖减少、穗粒数减少乃至小穗不孕等,严重影响燕麦的品质和产量,通常可造成30%~50%的产量损失[5,6],其已成为制约燕麦产业发展的主要病害[7,8]。麦长管蚜(Sitobion avenae)是燕麦产区的常发性蚜虫,也是燕麦红叶病的主要媒介昆虫。探讨BYDV 与麦长管蚜互作的分子机制,旨在为燕麦产业病虫害防控提供理论依据。
转录组是生物体特定组织或特定细胞在某一阶段产生的所有转录的集合,可用于比较该组织或细胞在不同处理条件下的基因表达差异[9-11]。利用差异表达基因分析、基因挖掘及基因结构分析的研究,在对昆虫生命活动过程中相关基因、昆虫与其他生物互作等方面的研究中发挥重要作用[12,13]。本研究对麦长管蚜进行饲毒(获毒)处理,利用转录组对感染BYDV 后麦长管蚜的基因表达进行研究,以期从分子层面上探讨BYDV 与麦长管蚜的互作机制,为防治燕麦红叶病提供基础理论依据。
1 材料与方法
1.1 试验材料
在蚜虫发生初期,从宁夏银川市西夏区园林场燕麦种植区田间采集无翅麦长管蚜成蚜,收集所产若蚜,转移至温室防虫网内栽培的健康燕麦幼苗上饲养,在人工气候室内进行续代繁殖,进行PCR 检测,确保该麦长管蚜群体不携带大麦黄矮病毒。在燕麦红叶病盛发期,从田间采集带有燕麦红叶病典型症状的燕麦叶片,带回实验室内进行PCR 检测,确保采集的叶片携带BYDV。
1.2 饲毒(获毒)处理
将室内饲养的无毒麦长管蚜置于20 ℃人工气候箱中饥饿12 h。饥饿处理后,将无毒蚜虫分别置于装有健康燕麦叶片和带毒燕麦叶片的培养皿内,在20 ℃人工气候箱黑暗条件下饲毒72 h,使无毒蚜虫充分获毒。将获毒的蚜虫处理组及对照组蚜虫分别装入冻存管进行编号,每个冻存管由20 头蚜虫组成,每个处理设置3 个生物学重复,共计6 份样品,液氮冻存后置于-80 ℃超低温冰箱保存,用于RNA的提取。
1.3 总RNA 提取及转录组测序
总RNA 采用Trizol 法提取,采用1%琼脂糖凝胶电泳和Nanodrop 2000 分光光度计分析RNA 降解程度和纯度,使用Agilent 2100 检测RNA 完整性,委托北京组学生物科技有限公司采用Illumina HiSeq 测序平台对获毒和未获毒的麦长管蚜样本进行转录组测序。
1.4 测序数据分析
测序得到的原始数据过滤,去除接头序列以及低质量数据,获得高质量数据,利用Hisat2 将高质量数据与参考基因组进行序列比对,获得注释信息。使用BLAST 软件将获得的unigenes 与NR、Swiss-Prot、GO、COG、KOG、Pfam、KEGG 七大数据库比对,获得unigenes 的功能分类信息。
利用DEseq2 方法进行样品间的差异表达分析,然后对差异检验的P-value 作多重假设检验校正,通过控制FDR来决定P-value 的阈值,得到差异检验的FDR值,根据基因的表达量计算该基因在不同样本间的差异表达倍数。将|Fold Change|≥1.5 且FDR<0.01 作为筛选标准,对筛选获得的差异表达的基因进行KEGG 和GO 功能分析。
2 结果与分析
2.1 测序数据分析
利用Illumina HiSeq 测序平台进行转录组测序,测序数据质量情况见表1。由表1 可知,获毒组和对照组6 份样品共获得51.57 Gb 的高质量数据,各样本平均产出超过7.07 Gb 的高质量数据,Q20 均在97.76%及以上,Q30 均在92.69%及以上,表明该测序结果质量高,可以用于后续分析。
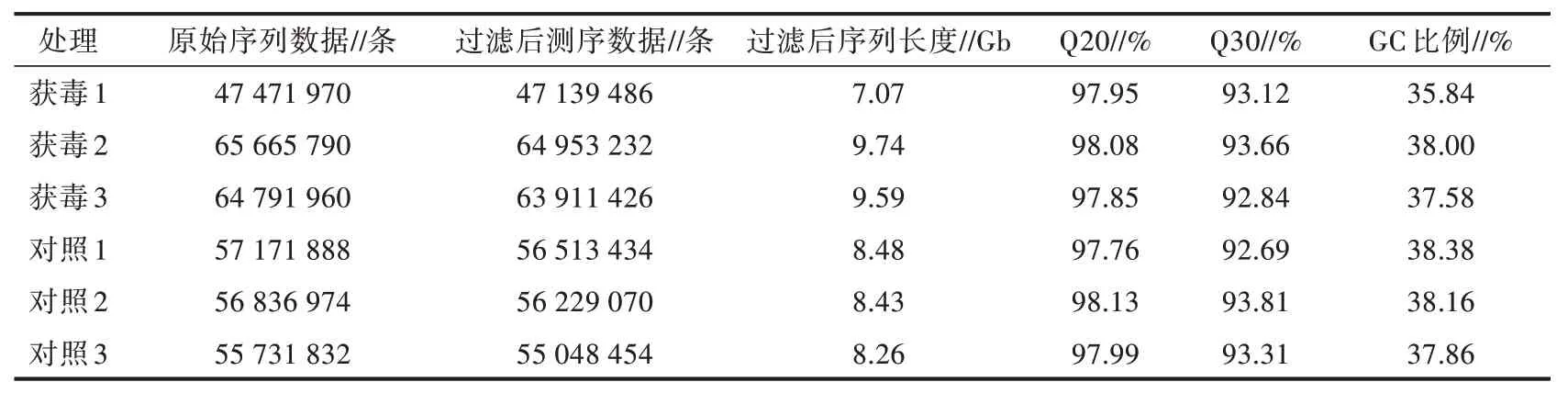
表1 测序数据质量情况
对预测的unigenes 进行基因注释,分别与七大数据库比对分析,结果见表2。由表2可知,最终获得28 798 条有注释信息的unigenes;11 342 条unigenes注释到KEGG 数据库的267 个代谢通路上;8 422 条unigenes 比对上KOG 数据库,并被分为25 个功能类别;10 452 条unigenes 被成功注释到GO 数据库中。

表2 七大数据库比对分析结果
2.2 差异表达基因分析
采用|Fold Change|≥1.5,FDR<0.01 为阈值筛选获毒蚜虫和未获毒蚜虫2 个转录本间的差异表达基因,结果见图1。由图1 可知,2 个转录本间共筛选到622 个差异表达基因,其中406 个表达上调,216 个表达下调。在筛选到的差异表达基因中,53 个差异基因能与NCBI的非冗余蛋白数据库比对并获得注释。
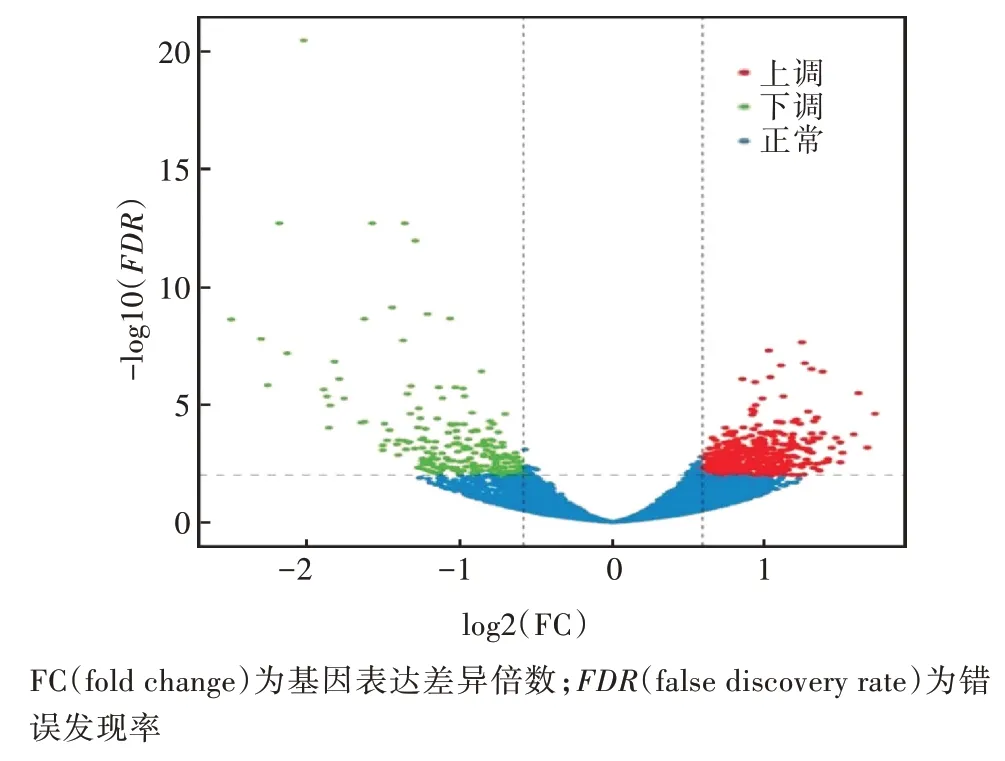
图1 获毒组与对照组麦长管蚜的差异基因火山
2.3 差异表达基因的GO 功能分析
对差异表达的基因进行GO 分析,36 个差异表达的基因被聚类到GO 的三大功能中,生物过程、分子功能和细胞成分中分别有30、53 和26 个基因,并且主要聚类在生物过程的代谢过程、细胞过程和生物调节;分子功能的结合和催化活性;细胞成分的细胞部分、细胞器和生物膜。筛选出20 个富集最显著的GO 功能,结果见图2。由图2 可知,在生物过程分类中,注释数量最多的是腺嘌呤核苷酸补救代谢,最低的是核苷酸代谢过程。在分子功能中显著性最高的为黄酮腺嘌呤二核苷酸结合,数量最低的为碱性磷酸酶活性。差异表达与代谢过程和催化活性相关的基因明显上调,说明麦长管蚜感染BYDV 后的代谢活动增强。

图2 差异表达基因的GO 富集
2.4 差异表达基因的KEGG 功能分析
对差异表达的基因进行KEGG 分析,有32 个差异表达的基因被注释到KEGG 的77 个代谢通路中。通过进一步统计分析代谢通路,筛选出前20 条富集最显著的通路(图3),主要有肌动蛋白细胞骨架的调节、PI3K-Akt 信号通路、轴突的导向、AMPK 信号通路和Ras 信号通路等。其中,差异基因注释到通路数量最多的是轴突导向,其次是PI3K-Akt 信号通路。筛选出8 个与免疫相关的差异表达基因,分别参与细胞免疫相关的溶酶体途径、吞噬体途径、MAPK 信号途径、Toll 样受体信号途径、Jak-STAT 信号途径以及TGF-beta 信号途径等,其中6 个基因的表达量上调。
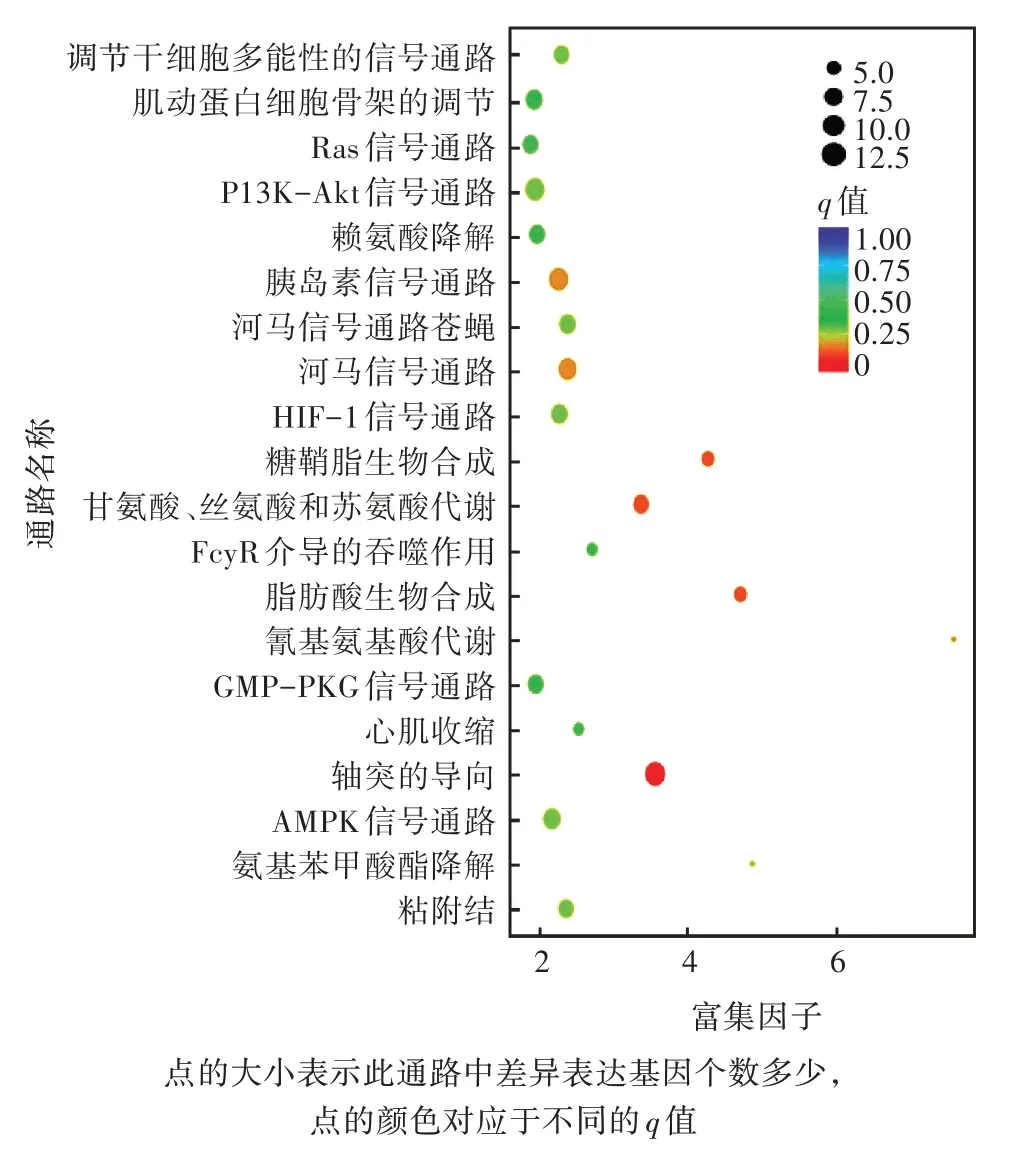
图3 差异表达基因的KEGG 富集
3 小结
本研究发现,带毒蚜虫催化活性相关的基因表达高于未带毒蚜虫。病毒在入侵宿主过程时会消耗大量的物质和能量,因此BYDV 可能会通过调节麦长管蚜体内的物质代谢活动,为BYDV 的入侵和增殖提供营养物质和能量。
本研究发现获毒后的麦长管蚜体内与溶酶体、吞噬体等途径相关的基因表达明显上调。细胞自噬是1 个重要的昆虫抗病毒免疫途径[14],这表明细胞自噬在麦长管蚜中可能起抗病毒的作用。
MAPK 信号途径、Toll 样受体信号路径、Jak-STAT 信号途径和TGF-beta 信号途径都参与昆虫的免疫反应[15-17]。带毒麦长管蚜体内与这些信号途径相关的基因的表达部分下调,说明BYDV 入侵麦长管蚜后,通过下调这些与免疫信号途径相关的基因抑制麦长管蚜的免疫反应。此外,这些免疫信号途径还参与了细胞增殖、分化等生长发育过程[18,19],病毒感染宿主媒介麦长管蚜后抑制了这些基因的表达,说明这些基因下调很可能降低麦长管蚜存活率[20]。
麦长管蚜对BYDV 的抵抗能力由多种代谢反应和信号转导途径共同调控。后续可以从转录组学检测到的代谢与信号转导通路入手,研究代谢与信号转导通路在麦长管蚜传毒的作用机制,为进一步研究BYDV 与蚜虫互作关系提供依据。此外,深入研究昆虫免疫系统能够提高农药控害效率[21],特别是对利用和开发生物农药进行害虫防治提供了重要的理论依据。
