A novel pathogenic splicing mutation of RPGR in a Chinese family with X-linked retinitis pigmentosa verified by minigene splicing assay
2023-10-21HuiQinWangPeiKuanCongTianHeXiaoFengYuYaNanHuo
Hui-Qin Wang, Pei-Kuan Cong, Tian He , Xiao-Feng Yu, Ya-Nan Huo
1Department of Ophthalmology, the Second People’s Hospital of Quzhou, Quzhou 324022, Zhejiang Province, China
2Key Laboratory of Growth Regulation and Translational Research of Zhejiang Province, School of Life Sciences,Westlake University, Hangzhou 310024, Zhejiang Province,China
3Department of Ophthalmology, Children’s Hospital of Hangzhou, Hangzhou 310005, Zhejiang Province, China
4Department of Ophthalmology, the Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou 310020,Zhejiang Province, China
Abstract
● KEYWORDS: retinitis pigmentosa; X-linked inheritance;RPGR; splicing mutation; pSPL3 minigene assay
INTRODUCTION
Retinal devolution leading to progressive visual loss can be caused by the common inherited disease, retinitis pigmentosa (RP, OMIM#226800)[1-2].Clinically, RP includes abnormalities of visual acuity, visual field constriction,night blindness, and eventually central visual loss[3-4].The worldwide prevalence of RP is approximately from 1/7000 to 1/3000 worldwide and is about 1/4000 in China[5-6].RP is a multiple inheritance pattern disorder.To date, more than 80 gene mutations have been identified in RP.This disease can be inherited in X-linked (XLRP; 5%-15%), autosomal recessive(50%-60%), and autosomal dominant (30%-40%) manners[7-8].RP is a highly heterogeneous form of inherited retinal degeneration, mainly affecting the rod photoreceptors, and XLRP is the most severe subtype[9].Male patients suffering from XLRP usually show a more severe phenotype than female carries[1,8].XLRP characteristically results in earlyonset night blindness and progressive peripheral vision loss,frequently leading to male sufferers becoming legally blind by 30 to 40 years old[10-11].Female carriers show a wider spectrum of phenotypes, from asymptomatic to severe[12].Presently,mutations in theRP2gene (encoding RP2 activator of ARL3 GTPase; OMIM# 300757), theOFD1gene(encoding Oral-Facial-Digital Syndrome 1, also known as OFD1 centriole and centriolar satellite protein; OMIM#300170), and theRPGRgene (encoding RP GTPase regulator; OMIM#312610)have been identified as causative in XLPR[13-14].RPGRmutations are considered to cause 70%-80% of XLRP cases and are responsible for approximately 11% of all RP cases in Caucasian patients[11,15].
XLRP demonstrates markedly heterogeneous phenotypes(including interfamily heterogeneity) such as the extent of the damage to rods and cones, the rate of progression, clinical severity, and the age of onset[9].Genetic characterizations of RP are complicated.To date, 58 genes have been reported to be associated with RP.With the aid of the latest technologies,such as whole-exome sequencing (WES), more hidden genesis events and new genetic mutations of RP could be uncovered effectively.In the ClinVar database[16], 132 variants are classified as pathogenic or likely pathogenic, among which 11 variants are splice sites mutations.Therefore, in the present study, WES technology and minigene splicing assays were used to determine a previously unknownRPGRsplice mutation in a Chinese family with a history of XLRP.
SUBJECTS AND METHODS
Ethical ApprovalThis study was carried out according to the tenets of the Declaration of Helsinki for research that involves human participants.The Ethics Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine approved the study (permit No.2020-0100).Written informed consent was obtained from each subject or their guardian before any study procedure was carried out.
Proband and His Clinical AssessmentThe proband was a 6-year-old boy who visited our clinic because of decreased vision and progressive night blindness over the past two years.The proband was tested using electroretinogram(ERG), pattern-visual evoked potential (P-VEP), vision field assessment, fundus photography, intraocular pressure measurement, slit lamp examination, refractive error measurement, and best-corrected visual acuity (BCVA).Following mydriasis using tropicamide (Mydrin-P, Santen Pharmaceutical, Japan), the refractive error was measured.An Octopus 900 perimeter (Haag Streit Internatinal, Koeniz,Switzerland) was used to assess the visual field.A digital fundus camera VISUCAM 200 (Carl Zeiss Meditec AG, Jena,Thuringia, Germany) was used to acquire high resolution images of the fundus.An RETIport32 electrophysiological system (Roland Consult GmbH, Wiesbaden, Germany) was used to record ERG and P-VEP.Information regarding the family history was obtained through interviews with subjects and their family members.
Whole-exome and Sanger SequencingDNA was extracted from the peripheral blood of the proband (IV-1), his 2-yearold younger brother (IV-2), their parents (III-3 and III-4), and an affected male relative (his great uncle, II-2).The proband(IV-1) was evaluated using targeted WES analysis.Library preparation and targeted enrichment were performed using Agilent SureSelect Human All Exon enrichment kit following the manufacturer’s instruction (Agilent Technologies, Santa Clare, CA, USA).An Illumina Hiseq 4000 instrument(Illumina, San Diego, CA, USA) was used to sequence the library.The sequencing data was analyzed mainly using the Burrows-Wheeler Aligner (BWA)[17]and the Genome Analysis Toolkit (GATK)[18].The variants were annotated using ANNOVAR (ANNOtate VARiation)[19]and compared with the data from the in-house database, gnomAD and the 1000 Genome Project.The pathogenicity of the splice site mutation was predicted using two programs, including Human Splicing Finder (HSF v3.1)[20]and NNSPLICE v0.9[21].DNA samples from other four affected members of this family were analyzed for the presence of the mutation using Sanger sequencing.
Minigene Construction, Cell Transfection, and Reverse Transcription Polymerase Chain ReactionTo investigate the effect of the intron 6 donor splice site mutation(c.619+1G>C), we developed anin vitrostrategy.Primers were designed to amplify regions of intron 4, exon 5, intron 5, exon 6, and part of intron 6 from the DNA of the proband.The primers were designed using NCBI Primer-Blast software.TheRPGRprimers (F: TTATGGGGTACGGGATCACCA GAATTCCGTTCCATGCATTCTGTTCA and R: ACGGG ATCACCAGATATCTGGGATCCCATGGACAACCATG GCATTA) produced an amplicon of 2034 bp.Next, wildtype (WT) control and mutant mini genes were constructed using plasmid pSPL3 (Invitrogen Corporation, Carlsbad, CA,USA).EcoRI and BamHI endonucleases were used to digest empty pSPL3 at 37℃ for 4h.Polymerase chain reaction (PCR)amplifications were carried out in a reaction comprising 16.5 μL of H2O, 3 μL of GC enhancer, 3 μL of 10× buffer,0.5 μL of DNA polymerase, 2 μL of dNTPs, 1 μL of each primer and 3 μL DNA in a 30-μL reaction volume.The PCR reaction consisted of an initial denaturation at 95°C for 5min followed by 30 cycles of 30s at 95℃, 30s at 61℃, and 2min at 72℃; followed by final extension for 10min at 72℃.The amplicons were purified using an AxyGen Mag PCR Clean-Up Kit (Axygen, Union City, CA, USA).
The amplicons were then recombined into pSPL3 as follows:81 ng of cleaned-up amplicon and 121 ng of linearized pSPL3 vector were added to 4 μL with 5× CE Ⅱ buffer and and 2 μL of Exnase Ⅱ in a 20 μL volume and incubated at 37℃for 30min (ClonExpress Ⅱ, Vazyme Biotech Co., Ltd, Nanjing,China).

Figure 1 Clinical data of the proband and II-2 A: Fundus photographs of the proband indicating arteriolar attenuation and bone-spicule pigmentanion; B: The field of vision of the proband showed severe constriction in both eyes; C: Pattern-visual evoked potential (P-VEP) of the proband showed an extinguished amplitude of the P100-wave in the lefteye; D: Both the rod and cone electroretinogram (ERG) amplitudes were extinguished; E: Fundus photographs of II-2 showing pallor of the disc, arteriolar attenuation, and bone-spicule pigment.
CompetentEscherichia coliDH5α cells were transformed with the WT and mutant mini gene constructs, which were then isolated from the transformants, using a NucleoBond Xtra midi kit (MN, Dueren, Germany).COS7 cells were then transfected with the WT, mutant, and empty control vector using PolyJet reagent (SignaGen Laboratories, MD, USA).Twenty-four hours later, we extracted total RNA from the cells and subjected it to reverse transcription-polymerase chain reaction (RT-PCR;Primer script RT reagent, Takara, Dalian, China).Two pSPL3 primers (F: TTGTGGAGATGGGGGTGGAGATGG and R: ACACATGGCTTTAGGCTTTGATCCC) were used for amplification from the WT, mutant and empty vector.The PCR products for both the WT and mutant variants were subjected to 2.5% agarose gel electrophoresis, followed by Sanger sequencing.
RESULTS
Clinical FeaturesThe proband (Figures 1A, 2B IV-1) was a 6-year-old Chinese boy.The BCVA of the left eye was 0.4 logMAR units, with a spherical refractive error of -3.00 D, and the visual acuity of the right eye was 0.2 logMAR units without a residual refractive error.Funduscopic examination showed retinal arteriole attenuation and mid-peripheral retinal scattered bone-spicule pigmentation in both eyes (Figure 1A).The visual fields were severely constricted in both eyes of the proband(Figure 1B).The P-VEP result showed low amplitude in the left eye (Figure 1C).The ERG results showed extinguished amplitudes (Figure 1D).The proband was diagnosed as having RP according to these criteria.In addition, two individuals (II-2 and II-4) from the same family were diagnosed as having RP in their local hospital.Images of II-2’s fundus revealed pallor of the optic disc, blood vessel attenuation, and multiplied bonespicule pigmentation (Figure 1E).In both II-2 and II-4, disease onset comprised night blindness within the first 10y of life and they were completely blind in their early fourth decade.None of them showed significant extra-ocular findings.The family’s pedigree is shown in Figure 2A.
Whole-exome Sequencing and Mutation AnalysisThe proband’s DNA was subjected to WES and the average depth was about 120×.All genes associated with RP were analyzed.We found a potential pathogenic variant in theRPGRgene in the proband (IV-1; Figure 2B).The male only allele of gene in the X chromosome.Male carries (IV-1, II-2 and IV-2)are hemizygous type for variant c.619+1G>C.The variant c.619+1G>C, located at the exon-intron boundary in theRPGRgene, has never been reported in all known databases,including our Chinese database of whole genome sequencing of 4500 individuals.We confirmed the variant in the other family members (II-2 and IV-2) by Sanger sequencing and the mother (III-4) of the proband was found to be heterozygous for variant c.619+1G>C.In this family, the variant co-segregated with the disease phenotype.HSF analysis of the sequences surrounding the mutated region indicate that it would impair normal splice donor site function, leading to a frame-shift in the open reading frame (Figure 3A).HSF Donor site variation score from 87.24 to 60.1 (-31.11%).NNSPLICE predict that c.619+1G>C disrupt the original donor site (NNSPLICE score:0.92 to 0.00).

Figure 2 The pedigree of the affected family and the mutation were identified using Sanger sequencing analysis A: In the pedigree,individuals with retinitis pigmentosa (RP) are indicated using filled symbols, clinically unaffected subjects are indicated using open circles or squares.Presumed and obligate carrier females are indicated with a dotted circle.The numbers beneath the symbols represent the sibship numbers of the individuals.The arrow indicates the proband in the family (IV-1).Slashes indicate individuals who are deceased.B:Representative sequence chromatograms for IV-1 (mutant hemizygous type), III-3 (wild-type), III-4 (mutant heterozygote type), and IV-2(mutant hemizygous type), II-2 (mutant hemizygous type).RPGR gene: NM_000328.3.
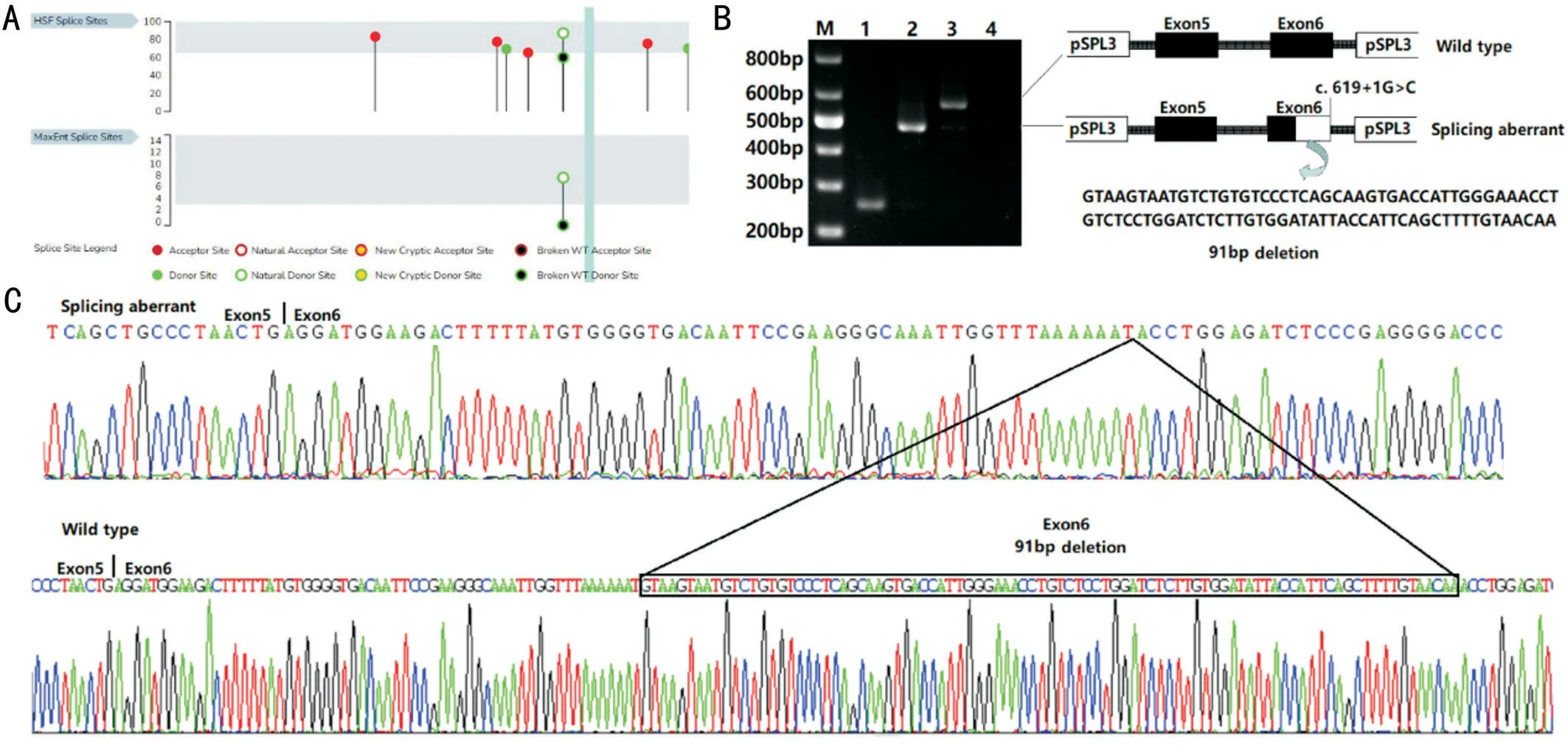
Figure 3 Minigene vectors constructs for the in vitro splicing assay A: Diagram of the minigene splicing assay.B: Reverse transcriptionpolymerase chain reaction (RT-PCR) products from c.619+1G>C in the pSPL3 minigene constructs.Lane M: DNA markers; Lane 1: empty vector;Lane 2: mutation; Lane 3: wildtype; Lane 4: blank control.C: Sanger sequencing of RT-PCR products.Mutation c.619+1G>C resulted in a 91 bp deletion in the end of exon 6.
Minigene AnalysisTo verify the pathogenicity of the splice site mutation, we constructed a minigene vectorinvitrosplicing assay for both the WT and mutant c.619+1G>CRPGRgene sequence from exon 5 to exon 6 (Figure 3).Upon RT-PCR,the WTRPGRexon 5 and exon 6 produced an amplicon of 572 bp (Figure 3A).The COS7 cells containing the empty vector (Figure 3A lane 1) produced a 263 bp band.The cells transfected with the c.619+1G>C mutation sequence produced a 481 bp band (Figure 3A).The mutation resulted in the activation of a cryptic splice site in the coding region of exon 6.Directing sequencing of the products showed that 91 bp (GT AAGTAATGTCTGTGTCCCTCAGCAAGTGACCATTGGG AAACCTGTCTCCTGGATCTCTTGTGGATATTACCATTC AGCTTTTGTAACAA) were deleted from the end of exon 6 in the mutated gene.
DISCUSSION
In the present study, we analyzed the genotype and phenotype of a Chinese family with RP.We found a new mutation,c.619+1G>C, located at the exon-intron boundary in theRPGRgene in the proband and other relatives (IV-1, II-2 and IV-2).The variant resulted in a partial deletion of the end of exon 6 (91 bp) and was predicted to cause aberrant mRNA splicing (p.Val177Glnfs*16).The variant was identified in the male patients (hemizygous), normal females (heterozygous)and absent in the healthy male members, which showed cosegregation in the family.To the best of our knowledge, the splicing mutation has not been reported in all known databases,including our Chinese database of whole genome sequencing data.The novel splice site variant c.619+1G>C of theRPGRgene was shown to be a pathogenic variant in this family.
Increasing numbers ofRPGRmutations have been revealed in recent years because of the widespread use of next-generation sequencing, which has also increased our knowledge concerning the spectrum of known phenotypes among patients.Most of the previously reported patients were recruited from Europe, North America, and Japan[22].
To date, more than 350 sequence variants have been described inRPGR.Systematic analyses from previous studies and using gnomAD showed that patients harboring mutations in exons 1 to 14 likely develop severe RP clinical features compared to patients with variants inORF15[23].The majority ofPRGRpathogenic variants are truncation variants.By contrast,missense and in-frame variants were more common in the RCC1-like domain and might be benign[24-25].In our study,the novel truncation variant is located on the intron 6 splice junction, resulting in a severe clinical phenotype in male patients at a very early age.In the family, II-2 and II-4 became total blind by their early forties.IV-1 had a two-year history of decreased vision and severely constricted visual field in both eyes at age 6.However, the female carrier, III-4, showed a normal phenotype except for aspherical refractive error of-3.00 D in both eyes.In addition, the six-year-old preschool proband visited us because of the diminution of vision.He had been diagnosed as myopic and had worn glasses for over one year after visiting another hospital.The proband exhibited myopia; therefore, specific RP screening was not performed initially until his parents mentioned that wearing glasses did not improve his vision at night and that two older male relatives were totally blind in their early forties.This reinforces that in high-risk families, performing comprehensive examinations of children at a young age, with or without myopia, is important.In cases where their vision did not improve after a period of myopia treatment, the possibility that RP or other diseases might exist should be considered for these patients.
In summary, we detected and identified a new pathogenic variant in theRPGRgene associated with XLRP with a severe phenotype in a Chinese RP family.The splice mutation c.619+1G>C causes aberrant splicing of exon 6 and intron 6,leading to a 91 bp deletion of the end of exon 6 in theRPGRgene, which resulted in very early onset of RP in the proband.The results revealed a furtherRPGRmutation that causes XLRP and added to the known range of phenotypes of this disease.Our findings could contribute to improved genetic consultation and diagnosis of XLRP.
ACKNOWLEDGEMENTS
Foundations:Supported by National Natural Science Foundation of China (No.31751003); Natural Science Foundation of Zhejiang Province (No.LY20H120009); Health Commission of Zhejiang Province (No.2022KY168); Beijing Bethune Charitable Foundation (No.BJ-GY2021013J).
Conflicts of Interest: Wang HQ,None;Cong PK,None;He T,None;Yu XF,None;Huo YN,None.
杂志排行

International Journal of Ophthalmology的其它文章
- A novel approach for 25-gauge transconjunctival sutureless vitrectomy to evaluate vitreous substitutes in rabbits
- Visual resolution under photopic and mesopic conditions in patients with Sjögren's syndrome
- Effects of obstructive sleep apnea on retinal microvasculature
- Bibliometric analysis of research relating to refractive cataract surgery over a 20-year period: from 2003 to 2022
- Three-dimensional bioprinting in ophthalmic care
- Agreement of intraocular pressure measurement with Corvis ST, non-contact tonometer, and Goldmann applanation tonometer in children with ocular hypertension and related factors