大肠杆菌混菌劳动分工发酵生产氨基葡萄糖
2023-10-18赵可欣耿自豪伊进行卓明洋张春月孙文超马倩
赵可欣,耿自豪,伊进行,卓明洋,张春月,孙文超,马倩
(天津科技大学 生物工程学院,天津,300457)
氨基葡萄糖(glucosamine, 2-amino-2-deoxy-D-glucose, GlcN),又称氨基葡糖、葡萄糖胺或葡糖胺,是一种重要的功能单糖,也是第一个被确认结构的氨基单糖[1-4]。氨基葡萄糖的传统生产方法主要为虾壳蟹壳酸水解提取,存在环境污染、易致敏等问题。近年来,随着代谢工程的迅速发展,微生物发酵法进行氨基葡萄糖生产的报道不断涌现[5],其中最主要的方法是对GlcN合成途径关键酶进行强化并阻断其分解利用的代谢途径。DENG等[6]通过易错PCR筛选出高效的氨基葡萄糖-6-磷酸合成酶基因(glmS),并将改良酶过量表达获得了氨基葡萄糖滴度17 g/L。董瑞真等[7-8]通过加强氨基葡萄糖合酶(fructose-6-phosphate transaminase,GlmS)和氨基葡萄糖乙酰化酶(glucosamine-6-phosphateN-acetyltransferase,Gna1)2种酶的表达,再对大肠杆菌氨基葡萄糖代谢途径进行改造,经摇瓶发酵验证后GlcN的产量提高至1.049 g/L。陈欣等[9]在已过表达GlmS和Gna1的大肠杆菌的基础上利用Red同源重组技术将乙酰氨基葡萄糖磷酸转运子编码基因nagE和甘露糖磷酸转运子编码基因manX敲除,在7 L发酵罐中发酵至10 h时GlcN产量达到最大值4.82 g/L。
目前利用微生物发酵直接合成GlcN的最终产量并不理想,主要是因为代谢过程中生成的氨基葡萄糖-6-磷酸对GlmS存在反馈抑制[10],且GlcN在发酵过程中不稳定易降解[6],因而GlcN在正常的代谢合成中不能大量积累。针对这一问题,工业发酵生产对GlcN进一步发酵生成N-乙酰氨基葡萄糖(N-acetyl-glucosamine,GlcNAc),之后再结合酸水解生成GlcN。但环境污染的问题仍未得到有效解决,同时增加了生产工艺复杂性,因此,需要创建一种直接高效生产GlcN的微生物发酵方法。
在长期的实验和生产实践中,人们不断地发现很多重要的生化过程必须依靠2种或多种微生物共同培养来完成[11]。如已广泛应用于生产的维生素C二步发酵法中的第二步发酵L-山梨糖转化形成2-酮基-L-古龙酸(维生素C前体物)是两菌混合发酵的过程[12]。混菌体系最大的特点是不同来源、不同功能的元件和模块可以在不同菌株中构建,既减轻对单菌底盘的代谢负荷,又便于将功能分区、避免功能间的交叉影响[13]。有研究在4株酵母菌中分别表达3种不同降解酶和一种蛋白支架,构成的混菌体系利用纤维素产乙醇的收率达到理论值的93%,解决了全部在单菌中构建导致代谢负担过重而无法工作的问题[14-15]。
本研究通过构建混菌发酵体系的方式直接生产GlcN,通过将GlcN的合成分为GlcNAc的高效合成与脱乙酰化2个步骤,并分别在2株大肠杆菌中进行代谢分工。将前期研究中构建的GlcNAc高效合成菌与本研究中构建的GlcNAc脱乙酰化菌组合进行混菌发酵,实现两菌劳动分工合成GlcN。
1 材料与方法
1.1 菌株、质粒及试剂
本研究所用的菌株与质粒信息详见表1。基因克隆与基因组编辑所用的引物信息见表2。所有的限制性内切酶、STAR HS DNA聚合酶、T4 DNA连接酶,TaKaRa公司;引物,苏州金唯智科技有限公司;N-乙酰氨基葡萄糖、氨基葡萄糖,Sigma-Aldrich。

表1 本研究所用菌株与质粒Table 1 Strains and plasmids used in this study

表2 本研究所用引物Table 2 Primers used in this study
1.2 大肠杆菌CRISPR/Cas9基因编辑
1.2.1 重组DNA片段构建
根据目的基因的序列,利用软件Primer Premier 5进行引物设计,通过PCR获得目的基因片段。以待整合或待敲除位点上下游序列为模板,设计合适的PCR引物,扩增500 bp左右的上、下游同源臂片段。对目的基因片段、上下游同源臂片段进行重叠PCR,以获得基因整合用的重组DNA片段。进行基因敲除时,则只对上下游同源臂片段进行重叠PCR构建。
1.2.2 pGRB质粒的构建
利用CRISPR RGEN Tools(http://www.rgenome.net/cas-designer/)选择待整合/敲除位点附近的PAM序列(5′-NGG-3′),并通过合成线性化引物将相应的20 bp的gRNA序列引入单链DNA中,之后通过PCR退火形成双链片段。将双链片段与线性化pGRB载体进行同源重组后转化至E.coliDH5α感受态中,筛选阳性克隆并提取质粒。
1.2.3 基因整合与敲除过程
参考LI等[16]报道的CRISPR/Cas9基因编辑技术进行E.coli基因组DNA的整合和敲除。
1.3 摇瓶发酵
培养基制备:
固体斜面培养基(g/L):蛋白胨10,酵母粉5,NaCl 10,牛肉膏10,琼脂粉20,pH 7.0~7.5。
种子培养基成分(g/L):葡萄糖20,酵母粉3,KH2PO42,柠檬酸2,MgSO4·7H2O 1,(NH4)2SO42,FeSO42.8 mg/L,MnSO41.2 mg/L,维生素B1 0.5 mg/L,维生素H 0.1 mg/L,微量元素混合液1 mL/L,消泡剂1滴,pH 7.0~7.2。
发酵培养基成分(g/L):葡萄糖20,酵母粉3,K2HPO46.67,(NH4)2SO44,柠檬酸3.55,谷氨酸2,MgSO4·7H2O 2.5,FeSO410mg/L,CaCl2·2H2O 25 mg/L,MnSO41.2 mg/L,NaCl 1,微量元素混合液1 mL/L,维生素B1 0.5 mg/L,维生素H 0.1 mg/L,消泡剂1滴。
首先在固体斜面培养基中进行菌种活化,置于37 ℃培养箱中培养12 h后转移至新的固体斜面,进一步培养12 h。用接种环刮取一环菌体,接种于装有30 mL种子培养基的500 mL圆底三角瓶中,用九层纱布封口,在37 ℃、200 r/min条件下培养10~12 h。
将培养好的种子培养液按接种后一定的OD600值比例,调整两菌接种体积,保持总接种量为10%(体积分数),接种于30 mL发酵培养基,在37 ℃、200 r/min条件下培养至发酵结束。在发酵过程中,根据培养基中苯酚红的颜色指示,通过补加氨水使发酵液pH始终维持在一定的范围。同时,根据耗糖情况,及时补加碳源。
1.4 发酵液中各成分的测定方法
本研究利用岛津液相色谱仪(SHIMADZU LC-20AT)采用HPLC对发酵液中GlcNAc、GlcN进行测定分析。GlcNAc的检测方法为色谱柱Aminex®HPX-87H色谱柱(7.8 mm×300 mm),检测器为RID-20A折光率检测器,流动相为5 mmol/L H2SO4溶液,柱温设定30 ℃,流速设定0.5 mL/min。GlcN的检测方法为参照DENG等[6]的检测方法。
样品检测:将待测样品13 000 r/min离心3 min,取上清液,稀释至合适浓度后用0.22 μm滤膜过滤,最后进行检测。
2 结果与分析
2.1 强化内源脱乙酰酶基因nagA对GlcN产量的影响
在前期的研究中构建了GlcNAc的高效合成菌GLA,该菌通过在E.coliW3110中引入酵母来源的gna1基因,过表达E.coli自身的glmS基因,同时敲除6-磷酸N-乙酰甘露糖胺差向异构酶编码基因nanE、乙酰氨基葡萄糖磷酸转运子编码基因nagE和甘露糖磷酸转运子编码基因簇manXYZ,阻断了GlcNAc的分解利用途径,实现了E.coli中GlcNAc的发酵生产[17]。如图1所示,GlcNAc的脱乙酰化可以通过2种方式实现:方案1,通过强化大肠杆菌内源性的脱乙酰酶基因nagA,同时敲除nanE、manXYZ和6-磷酸葡糖胺脱氨酶编码基因nagB,阻断了GlcNAc和GlcN的分解途径;方案2,通过引入不同来源的脱乙酰酶基因,择优选取后进一步强化,并对GlcNAc进入细胞的磷酸化途径进行阻断,同时强化胞内去磷酸化和阻断GlcNAc的分解途径。
为了验证混菌发酵直接生产GlcN的可行性,在出发菌E.coliDA中,敲除了nanE和nagB,得到了DAN菌株,再在DAN菌株的基础上对内源性的脱乙酰酶基因nagA进行不同拷贝数的强化,DAN-1、
DAN-2、DAN-3分别为nagA基因的单、双、三拷贝强化的菌株,并将上述菌株分别与GlcNAc合成菌株GLA进行摇瓶混菌发酵(菌种比例初步按体积比1∶1接种)。当菌株GLA、DAN、DAN-1进行单菌发酵时,发酵液中并无GlcN的产生,但当进行混菌发酵时,DAN+GLA的GlcN产量为3.6 g/L(图2-a),证明了混菌体系可以直接发酵生产GlcN。从图2-b中可看出,DAN-2+GLA的GlcN的产量最高为7.69 g/L,但当nagA基因拷贝数为3时,GlcN的产量开始下降,可能是因为由T7启动子控制的基因数量过多导致细胞负担加重,对GlcNAc的转化能力降低。
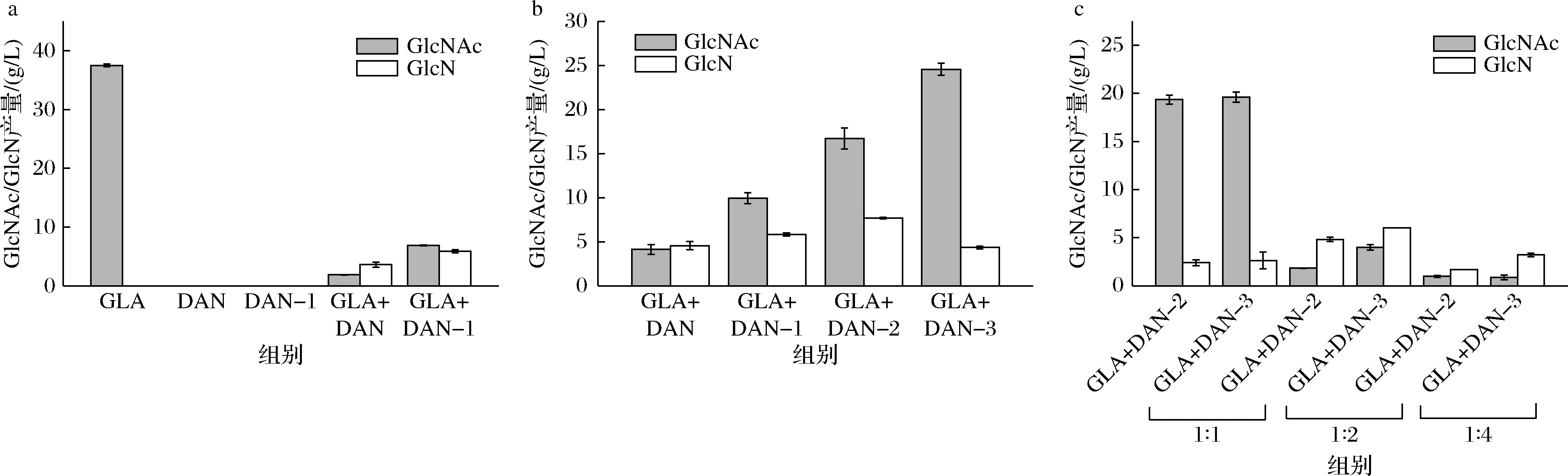
a-单、混菌发酵对GlcN产量的影响;b-nagA拷贝数对GlcN产量的影响;c-菌种比例对GlcN产量的影响
由于发酵液中不同的菌种比例会对产物合成产生较大影响,所以进一步对两菌接种比例进行了优化。从图2-c中可以看出,在OD600比例GLA∶DAN-3=1∶2时GlcN产量达到同批最高为6.03 g/L。
2.2 强化外源性脱乙酰酶基因对GlcN产量的影响
为了能获得更高的GlcN产量,本研究引入了来源于火球菌(Pyrococcushorikoshii)的二乙酰壳二糖脱乙酰酶基因phD[18]和来源于海洋环杆菌(Cyclobacteriummarinum)的脱乙酰酶基因cyD[19]。由于这2种外源酶催化底物为不带磷酸基团的GlcNAc,所以首先将DA菌株对GlcNAc进入细胞的磷酸化途径进行阻断得到了DAD菌株,再在DAD菌株的基础上分别整合phD和cyD基因,得到DAD-1和DAD-2菌株。为检验并对比上述2种脱乙酰酶的活性,在发酵培养基中添加GlcNAc使其浓度达到50 g/L,并对DAD、DAD-1、DAD-2进行单菌发酵,得到DAD-1产量最高为5.24 g/L(图3-a)。再将这3株菌分别与GLA进行混菌发酵,36 h后测得GLA+DAD-1中GlcN的产量最高为2.04 g/L(图3-a),故选择来源于火球菌的phD基因进行后续实验。
在DAD-1的基础上进行phD基因的强化,DAD-3、DAD-4分别为phD基因的双拷贝和三拷贝,将DAD、DAD-1、DAD-3、DAD-4分别与GLA进行接种比例为1∶1的混菌发酵,得到的产量分别为2.19、4.37、5.43 g/L(图3-b)。鉴于图2-c中接种比例为1∶4时GlcNAc和GlcN的产量均较低,所以此次比例优化调整为1∶1、1∶2、1∶3,且缩短发酵时间至24 h,以免长时间的发酵导致GlcN的持续降解。当GLA∶DAD-3=1∶1,发酵24 h时产量达到最高为7.83 g/L(图3-c),GlcN产量有了明显提高。为了防止拷贝数过多导致菌体生长和代谢受阻,选择在DAD-3的基础上做后续优化。
2.3 进一步优化组合对GlcN产量的影响
中性pH条件下,含有自由氨基的GlcN是不稳定的,而在酸性条件下较为稳定[20],所以在新的发酵条件,即菌种比例1∶1、低pH值(pH≤6.7)下发酵24 h后测定氨糖产量。GLA+DAD-3的GlcN产量达到9.69 g/L,相较图3-c时的最高产量提高了约1.23倍。研究表明大肠杆菌周质空间中含有几种磷酸酶[21],可能会使一小部分GlcNAc磷酸化进入细胞,从而降低转化率。为了进一步优化菌种,采用了2种方式菌种进行优化:强化胞内磷酸化基因yqaB(卤酸脱卤酶样磷酸酶)[22-23],使GlcNAc-6P去磷酸化生成GlcNAc,进而脱乙酰化生成GlcN;强化内源性脱乙酰酶基因nagA,分2种脱乙酰化途径合成GlcN。对DAD-5(Trc-nagA)和DAD-6(Trc-yqaB)进行摇瓶发酵得到GlcN的产量为9.02和11.21 g/L。GLA+DAD-6比同批发酵的GLA+DAD-3提高了1.15倍,较初始对照体系GLA+DAN提高了约3.1倍(图4)。

图4 强化胞内磷酸化基因yqaB和内源脱乙酰酶基因nagA对GlcN产量的影响Fig.4 Effect of the enhancement of intracellular phosphorylation gene yqaB and the endogenous deacetylase encoding gene nagA on GlcN titer
3 讨论
本研究采用混菌发酵的方法,成功地将GlcN的合成分为GlcNAc的高效合成与脱乙酰化2个步骤,并分别在2种大肠杆菌中进行代谢分工。在构建GlcNAc脱乙酰化菌的过程中,通过强化大肠杆菌内源性的脱乙酰酶基因nagA来增强菌株对GlcNAc的脱乙酰化能力,得到最高GlcN产量为7.69 g/L;为了进一步提高GlcN的产量,引入不同来源的脱乙酰酶基因并进行比较,优化基因拷贝数和菌种比例,并改变发酵条件防止产物降解严重,最终确定最优菌株和发酵条件,得到GlcN产量为7.83 g/L;为了提高GlcNAc的转化率,在最优条件的基础上强化胞内去磷酸化得到GlcN 11.21 g/L,较初始对照体系(GLA+DAN)提高了约3.1倍,此时的GlcN的转化率为0.09 g/g葡萄糖,GlcNAc脱乙酰化的效率达到了70.46%,较低于文献中所报道的转化率86.8%[18]。可能是因为混菌体系中代谢途径较为复杂,引入的脱乙酰酶的最适反应条件与发酵条件可能存在一定的差异。由此可见,在本研究构建的混菌体系中,对碳源的利用率还有待提高,且进一步提高细胞内的脱乙酰化效率也有利于GlcN产量的提升。
