断奶马驹小肠菌群多样性分析
2023-10-16李佳豪陈开旭李晓斌
李佳豪 李 倩 陈 晖 陈开旭 李晓斌*
(1.新疆农业大学,新疆肉乳用草食动物营养实验室,乌鲁木齐 830052;2.新疆农业大学,新疆马繁育与运动生理重点实验室,乌鲁木齐 830052)
动物肠道中栖居着大量的微生物,这些微生物直接参与宿主的诸多生理活动,包括促进消化道发育、提高宿主对饲粮中营养物质的消化吸收、减缓肠道应激和提高肠道免疫等[1-3]。马属动物小肠主要以化学消化为主,是蛋白质、非结构性碳水化合物和脂肪消化的主要场所。小肠中也有丰富的微生物,十二指肠和空肠的细菌数量为2.90×106CFU/g,回肠的细菌数量为38.4×106CFU/g[4]。研究表明,马消化道相邻区段的菌群多样性和丰富度存在差异,而这种差异一定程度上可能是导致宿主肠道消化、生理等机能差异的原因[5]。断奶应激会导致动物胃肠道组织结构和功能发生剧烈变化,进而影响动物的生长发育[6]。因此,了解断奶后幼龄动物肠道菌群的多样性和丰富度,对于调控幼龄动物肠道菌群结构和缓解断奶应激具有重要意义。目前,关于马肠道菌群的研究主要集中在粪便[7-8],对马不同肠段中菌群结构和种类的研究鲜见。粪便只能代表后肠道远端区域的菌群变化[9],而不能反映整个肠道菌群的变化。因此,本试验以5月龄断奶哈萨克马驹为研究对象,通过高通量测序技术检测其十二指肠、空肠和回肠菌群的多样性,探究马消化道相邻区段的菌群结构和多样性,以期为全面评价断奶后马属动物小肠发育、健康、功能等提供参考依据。
1 材料与方法
1.1 试验动物
在新疆昭苏县喀拉苏镇兴昭牧业场,选择出生日期(2019年3月出生,±5 d)和断奶日期(5月龄)相近的哈萨克马公马驹6匹为研究对象。
1.2 试验设计
6匹5月龄断奶哈萨克马公马驹在相同的饲养管理和饲粮营养水平条件下,进行为期60 d的饲养试验。6匹断奶马驹在第60天进行屠宰,分别采集十二指肠、空肠和回肠的内容物,采用 16S rRNA高通量测序技术检测各肠段内容物中菌群的多样性,并比较分析3个肠段菌群组成的差异性。试验开始时马驹平均体重为(112.36±7.50) kg,屠宰时平均体重为(147.08±4.86) kg。
1.3 饲养管理
所有马匹在试验前按照0.2 mg/kg BW的剂量口服伊维菌素(ivermectin)驱虫剂进行驱虫。每日09:00—21:00马驹室外活动饲养,将全天的精料补充料平均分成3等份,分别在09:00、15:00和21:00使用料兜进行饲喂。马驹在活动场(80 m×40 m)自由活动,单槽位自由采食粗饲料(苜蓿和干草2∶1均匀混合饲喂)。当天21:00至第2天09:00马驹室内单圈舍饲养(4 m×3 m,内置垫草),并提供粗饲料和清洁的饮水。精料补充料组成及营养水平见表1。

表1 精料补充料组成及营养水平(干物质基础)
1.4 样品采集
经过新疆农业大学实验动物伦理委员会同意后,将禁食12 h的马驹屠宰后,参照解剖学方法,分离十二指肠、空肠和回肠,立刻将内容物放置在高压灭菌的玻璃烧杯中,用玻璃棒混合均匀,随机取5 g装于无菌无RNA的冻存管中,液氮速冻后放置于-80 ℃冰箱中冷冻保存待测。
1.5 菌群多样性高通量测序分析
DNA提取、PCR扩增、Illumina HiSeq测序及结果分析均由北京诺禾致源生物信息科技有限公司协助完成。
1.5.1 样品基因组DNA提取及PCR扩增
取大约0.25 g肠道内容物样品,采用十六烷基三甲基溴化铵(CTAB)法提取样本的基因组DNA,用0.8%的琼脂糖凝胶电泳检测纯度和浓度,稀释至1 ng/μL备用。以稀释后的基因组DNA为模板,根据测序区域选择V3~V4区特异引物338F(5′-ACTCCTACGGGAGGCAGCA-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)进行PCR扩增。
1.5.2 文库构建和测序
使用NEBNext®UltraTMIIDNA Library Prep Kit建库试剂盒进行文库构建,将构建好的文库进行Qubit和PCR定量;待文库合格后,使用NovaSeq 6000测序仪进行测序。
1.5.3 测序数据处理
从下机数据中拆分出各样本数据,然后截去Barcode和引物序列用于拼接,得到的拼接序列即为原始数据。参照Qiime的质量控制流程,将原始数据进行截取,并过滤掉其中连续高质量碱基长度小于序列长度75%的数据。将经过以上处理后得到的序列与数据库进行比对,检测并去除其中的嵌合体序列,得到最终的有效数据。
1.6 数据处理
1.6.1 alpha多样性数据处理
使用Qiime软件计算Observed_species指数、Chao1指数、Shannon指数、Simpson指数、ACE指数、覆盖度(Goods-coverage)和PD_whole_tree指数。使用R软件绘制稀释曲线、Rank abundance曲线、物种累积曲线。使用R软件进行alpha多样性指数组间差异分析,并选用Tukey检验对alpha多样性指数组间差异分别进行有参数检验和非参数检验。
1.6.2 beta多样性数据处理
使用Qiime软件计算Unifrac距离,构建UPGMA样本聚类树。使用R软件绘制主成分分析(PCA)和主坐标分析(PCoA)图。PCA使用R软件的ade4和ggplot2软件包,PCoA使用R软件的WGCNA、stats和ggplot2软件包。使用R软件进行beta多样性指数组间差异分析,并选用Tukey检验对beta多样性指数组间差异分别进行有参数检验和非参数检验。
LEfSe分析使用LEfSe软件,默认设置线性判别分析得分(LDA score)的筛选值为4。Metastats分析使用R软件在各分类水平下做组间的排列检验(permutation test),得到P值,然后利用Benjamini and Hochberg False Discovery Rate方法对P值进行修正。组间差异显著的物种分析利用R软件做组间Tukey检验。
1.6.3 功能注释
Tax4Fun功能预测是通过基于最小16S rRNA序列相似度的最近邻居法实现,其具体做法为提取KEGG数据库原核生物全基因组16S rRNA基因序列并利用BLASTN算法将其比对到SILVA SSU Ref NR数据库(BLAST bitscore>1 500)建立相关矩阵,将通过UProC和PAUDA方法注释的KEGG数据库原核生物全基因组功能信息对应到SILVA数据库中,实现SILVA数据库功能注释。测序样品以SILVA数据库序列为参考序列聚类出操作分类单元(OTUs),进而获取功能注释信息。
2 结果与分析
2.1 十二指肠、空肠和回肠菌群alpha多样性分析
由表2可知,断奶马驹十二指肠、空肠和回肠菌群的Observed_species指数、Shannon指数、Simpson指数和Chao1指数均无显著差异(P>0.05),但十二指肠菌群的ACE指数和PD_whole_tree指数均显著高于空肠(P<0.05)。各肠段覆盖度均超过0.99,说明数据均能准确反映断奶马驹十二指肠、空肠和回肠菌群的组成。

表2 断奶马驹十二指肠、空肠和回肠菌群alpha多样性分析
2.2 基于OTUs的韦恩图
韦恩图(图1)显示了3个肠段之间的共有OTUs数量以及各肠段独有的OTUs数量。3个肠段共有的OTUs数量为1 153个,十二指肠特有的OTUs数量为550个,空肠特有的OTUs数量为679个,回肠特有的OTUs数量为281个。空肠特有的OTUs数量高于十二指肠和回肠,这表明在空肠中检测到更多的细菌种类。
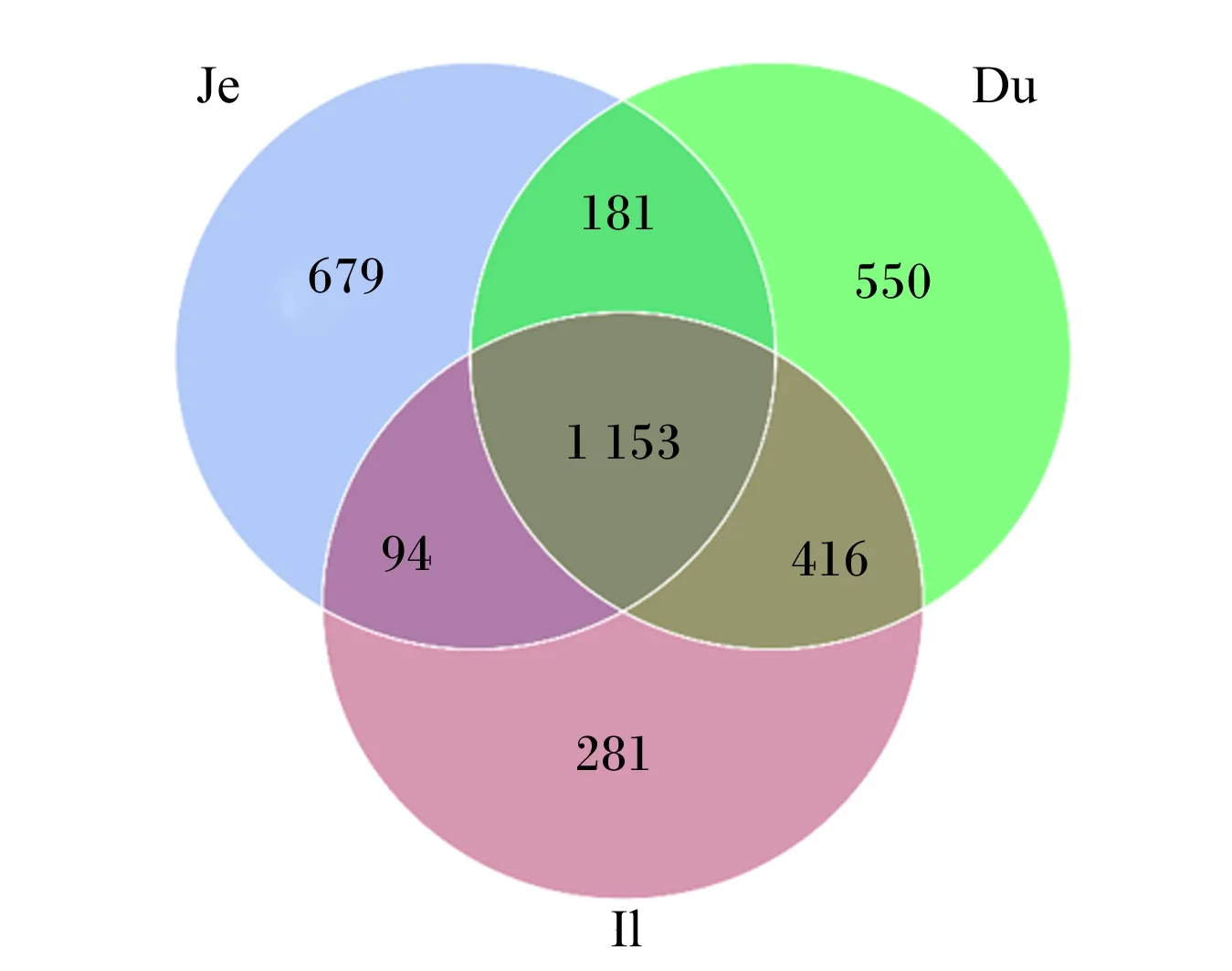
Du、Je和Il分别表示十二指肠、空肠和回肠。下图同。
2.3 十二指肠、空肠和回肠菌群beta多样性分析
十二指肠、空肠和回肠菌群beta多样性分析见图2。由PCoA图可知,第1主成分对样本变异的贡献为24.61%,第2主成分对样本变异的贡献为12.10%,十二指肠和回肠菌群的相似性较高;PCA图显示,第1主成分对样本方差的贡献为17.55%,第2主成分对样本方差的贡献为9.98%,十二指肠、空肠和回肠菌群的相似性较高。
2.4 十二指肠、空肠和回肠菌群门水平物种丰度分析
由表3可知,断奶马驹十二指肠、空肠和回肠菌群在门水平上相对丰度排在前10的物种为厚壁菌门、变形菌门、拟杆菌门、放线菌门、蓝藻菌门、酸杆菌门、螺旋体门、梭杆菌门、芽单胞菌门和疣微菌门,其中厚壁菌门、变形菌门和拟杆菌门的相对丰度之和在98%以上。空肠菌群中厚壁菌门的相对丰度比回肠提高了26.98%,差异显著(P<0.05);回肠菌群中变形菌门和梭杆菌门的相对丰度显著或极显著高于十二指肠和空肠(P<0.05或P<0.01)。
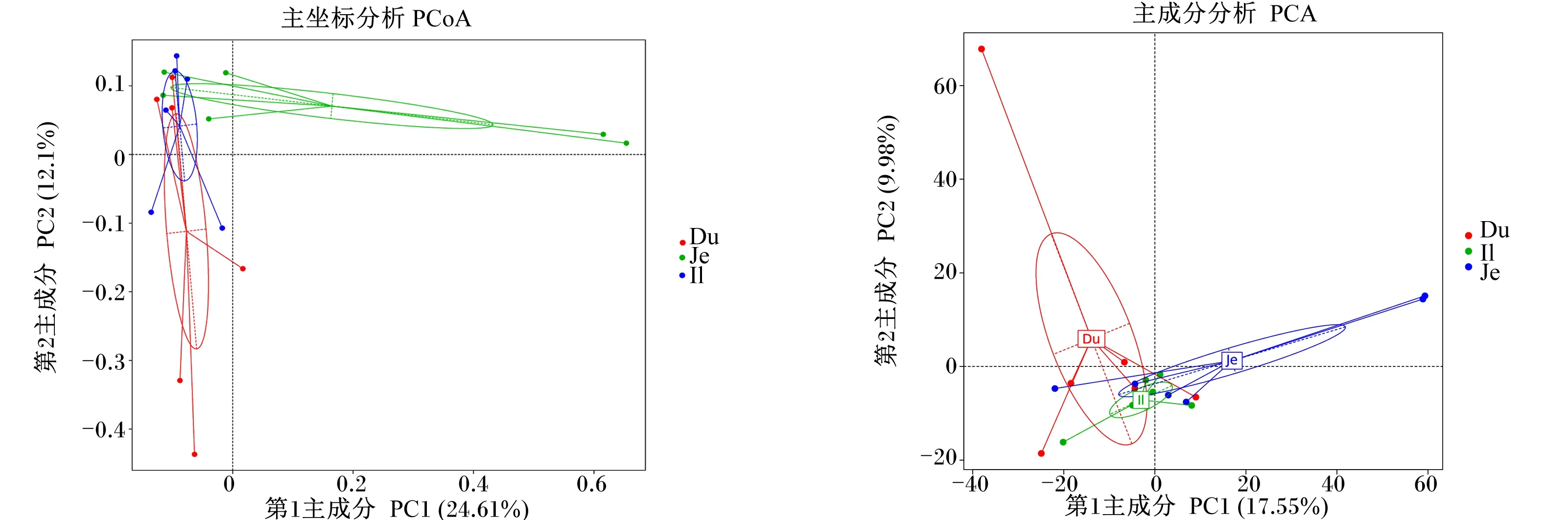
图2 十二指肠、空肠和回肠菌群beta多样性分析

表3 断奶马驹十二指肠、空肠和回肠内容物菌群门水平物种丰度分析
2.5 十二指肠、空肠和回肠菌群科水平物种丰度分析
由表4可知,断奶马驹十二指肠、空肠和回肠菌群在科水平上相对丰度排在前10的物种为链球菌科、乳杆菌科、肠杆菌科、巴斯德氏菌科、梭菌目未鉴定科、毛螺菌科、瘤胃球菌科、瘤胃球菌科、普氏菌科、韦荣菌科和黄色杆菌科。回肠菌群中肠杆菌科和巴斯德氏菌科的相对丰度均显著高于空肠(P<0.05),且巴斯德氏菌科的相对丰度还显著高于十二指肠(P<0.05);十二指肠菌群中瘤胃球菌科的相对丰度较空肠提高了156.04%,差异显著(P<0.05)。
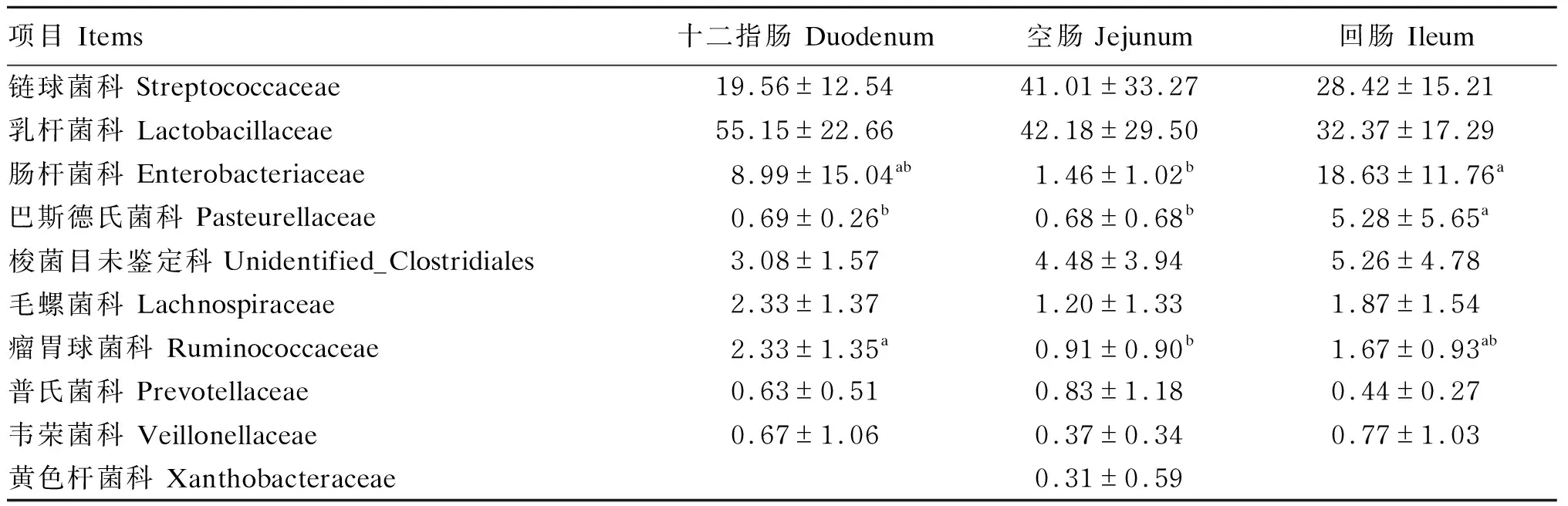
表4 断奶马驹十二指肠、空肠和回肠菌群科水平物种丰度分析
2.6 十二指肠、空肠和回肠菌群属水平物种丰度分析
由表5可知,断奶马驹十二指肠、空肠和回肠菌群在属水平上相对丰度排在前10的物种为乳酸菌属、链球菌属、肠杆菌科未鉴定属、放线杆菌属、梭菌目未鉴定属、乳球菌属、韦荣氏球菌属、八叠球菌属、柠檬酸杆菌属和红假单胞菌属。回肠菌群中肠杆菌科未鉴定属和放线杆菌属的相对丰度均显著高于空肠(P<0.05),且放线杆菌属的相对丰度还显著高于十二指肠(P<0.05)。
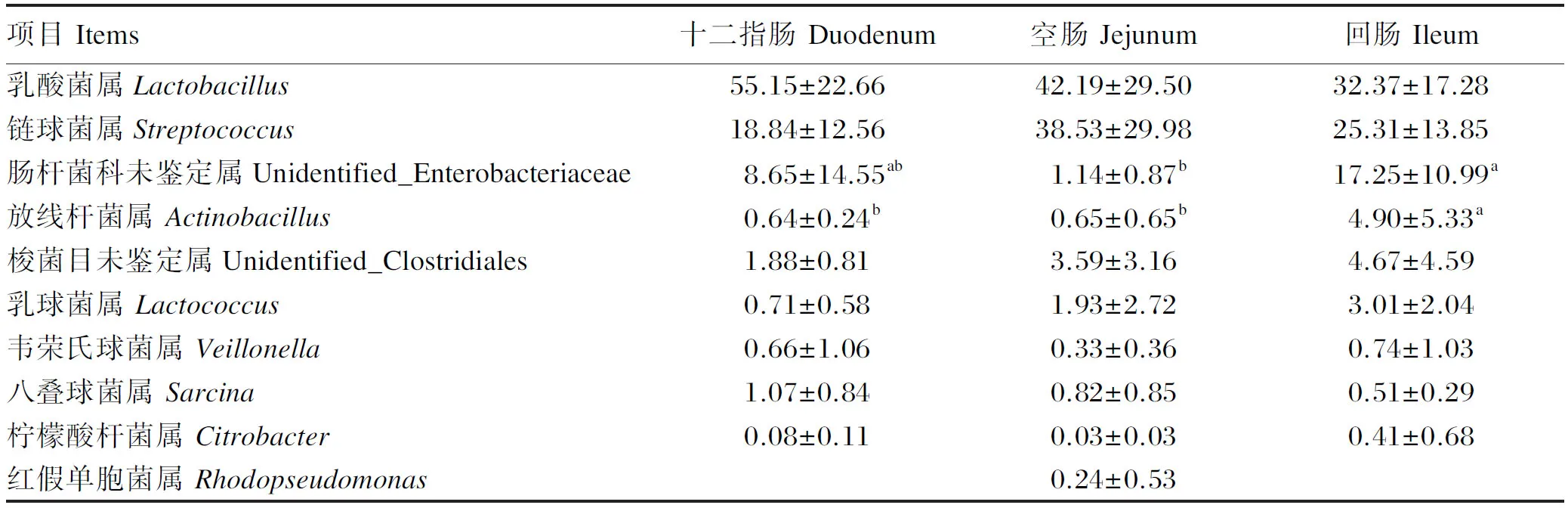
表5 断奶马驹十二指肠、空肠和回肠菌群属水平物种丰度分析
2.7 十二指肠、空肠和回肠菌群LEfSe分析
LEfSe分析能够在组与组之间寻找有统计学差异的物种。由图3可知,空肠与回肠菌群中有显著性差异的物种有10个,其中空肠菌群中有显著差异的物种有4中,分别为芽孢杆菌纲、乳杆菌目、厚壁菌门和细菌界;回肠内有差异的物种有6中,分别为大肠埃希氏菌、肠杆菌科未鉴定属、肠杆菌科、肠杆菌目、变形菌门和γ-变形菌纲。

k_:界 kingdom;p_:门 phylum;c_:纲 class;o_:目 order;f_:科 family;g_:属genus;s_:种 species;Bacilli:芽孢杆菌纲;Lactobacillaes:乳酸杆菌目;Firmicutes:厚壁菌门;Bacteria:细菌界;Escherichia coli:大肠埃希氏菌;unidentified_Enterobacteriaceae:肠杆菌科未鉴定属;Enterobacteriaceae:肠杆菌科;Enterobacteriale:肠杆菌目;Proteobacteria:变形菌门;Gammaproteobacteria:γ-变形菌纲。
2.8 十二指肠、空肠和回肠菌群Tax4Fun功能预测
由图4可知,断奶马驹十二指肠、空肠和回肠菌群的功能差异明显,其中空肠菌群主要为酶家族(enzyme families)、脂质代谢(lipid metabolism)、外源性生物降解和代谢(xenobiotics biodegradation and metabolism)、细胞群落原核生物(cellular community prokaryotes)、其他氨基酸的代谢(metabolism of other amino acids)、萜类及酮类化合物代谢(metabolism of terpenoids and polyketides)以及折叠、分类和降解(folding, sorting and degradation)等功能;回肠菌群主要为辅因子和维生素的代谢(metabolism of cofactors and vitamins)、膜运输(membrane transport)、能量代谢(energy metabolism)和翻译(translation)等功能;十二指肠菌群主要为转录(transcription)和碳水化合物代谢(carbohydrate metabolism)等功能。
3 讨 论
马属动物胃肠道拥有丰富多样的微生物群落,这些微生物群落通过降解和发酵饲粮中碳水化合物为宿主提供必需的营养物质[2,10];同时,微生物群落在调控机体免疫和维持宿主健康等方面也具有重要作用[11]。相对于反刍动物而言,单胃草食动物主要在盲肠和结肠利用微生物进行纤维的发酵和降解[12],小肠则更多的是将易消化的碳水化合物如淀粉、糖等分解为更小的分子后吸收进入血液循环[13]。因此,目前关于马属动物肠道微生物的研究更多的集中在盲肠和粪便中,对于其小肠微生物的研究鲜见。Dicks等[14]研究表明,马小肠内的微生物数量众多,活菌数在106~107CFU/mL。徐娥等[15]通过探究大约克猪肠段不同部位微生物相对丰度和多样性发现,十二指肠、空肠和回肠内容物中细菌总数呈现回肠>空肠>十二指肠,且各肠段菌群的丰富度和多样性随着消化道的深入呈现逐渐增加的趋势。朱春红等[16]为了揭示鸭肠道菌群特征,以健康的高邮鸭为研究模型,通过高通量测序发现,健康鸭十二指肠、空肠以及回肠菌群中OTUs数量均有所差异,且OTUs的数量呈现出空肠>十二指肠>回肠。本研究结果显示,断奶马驹十二指肠、空肠及回肠菌群共有的OTUs数量为1 153个,随着肠道结构的不断深入,各肠段菌群中OTUs数量呈现出空肠>十二指肠>回肠。十二指肠、空肠及回肠菌群的多样性指数分析结果显示,十二指肠和回肠菌群多样性均高于空肠,且十二指肠与空肠呈现显著关系。PCA结果显示,十二指肠、空肠和回肠物种组成相似。造成这种差异的原因可能与各肠段的生理状态和功能有关。十二指肠与胃相连,其酸性内容物以及腺体分泌的胰液、胆汁等创造的消化性环境不利于微生物的生存[17],而空肠是马属动物小肠中最长的部分,内容物周转相对缓慢,可能更适于微生物的生存。

Metabolism:代谢;Enzyme families:酶家族;Lipid metabolism:脂质代谢;Xenobiotics biodegradation and metabolism:外源性生物降解和代谢;Cellular community prokaryotes:细胞群落原核生物;Metabolism of other amino acids:其他氨基酸的代谢;Metabolism of terpenoids and polyketides:萜类及酮类化合物代谢;Folding, sorting and degradation:折叠、分类和降解;Poorly characterized;缺失的功能描述;Amino acid metabolism:氨基酸代谢;Drug resistance:药物抵抗;Cellular processes and signaling:细胞过程和信号;Cell motility;细胞运动;Transport and catabolism:运动和分解;Genetic information processing:遗传信息处理;Transcription:转录;Signal transduction:信号转导;Carbohydrate metabolism:碳水化合物代谢;Glycan biosynthesis and metabolism:糖的生物合成与代谢;Metabolism of cofactors and vitamins:辅因子和维生素的代谢;Membrane transport:膜运输;Energy metabolism:能量代谢;Translation:翻译;Nucleotide metabolism:核苷酸代谢;Replication and repair:复制与修复。
由于肠道组织结构、生理功能及饲粮结构的不同,不同肠段的菌群结构和功能存在明显差异[18-19]。Dougal等[20]通过16S rDNA测序技术对10匹纯种马和英国本土马的回肠和大肠内容物进行检测发现,所有马匹的回肠样品中隶属于厚壁菌门的乳杆菌科和隶属于变形菌门的巴氏杆菌科的相对丰度最高。吾尔恩·阿合别尔迪等[21]指出,马回肠优势菌为放线菌属和狭义梭菌属。研究显示,厚壁菌门内的细菌多数与碳水化合物代谢有关,大多数的厚壁菌门细菌能够分泌胞外多糖降解酶[22-23],而变形菌门内的细菌多数与蛋白质发酵有关[22-24]。本研究发现,断奶马驹十二指肠菌群中瘤胃球菌科的相对丰度显著增加,空肠菌群中厚壁菌门的相对丰度显著增加,回肠菌群中变形菌门、梭杆菌门、肠杆菌科、巴斯德氏菌科、肠杆菌科未鉴定属和放线杆菌属的相对丰度显著增加,这与Li等[25]和李倩等[26]分析断奶马驹胃和盲肠中菌群结构所得结果相符合。饲粮中的非结构性碳水化合物在马属动物胃和小肠(十二指肠、空肠及回肠)中被消化吸收,进而导致小肠中厚壁菌门和变形菌门的相对丰度较高。随着饲粮的非淀粉多糖在十二指肠、空肠和回肠中逐渐降解为可溶性寡糖,为盲肠中降解纤维类物质的细菌提供了适宜的可利用底物。因此,断奶马驹盲肠中拟杆菌门的相对丰度上升,而厚壁菌门和变形菌门的相对丰度下降。
为了进一步研究断奶马驹不同肠段菌群组成和物种丰度的差异,本试验对不同肠段菌群进行了LEfSe分析,发现空肠与回肠菌群中存在显著差异的物种有10个,其中空肠菌群中有显著差异的物种有4个,分别为细菌界、厚壁菌门、芽孢杆菌纲和乳酸杆菌目;回肠内有显著差异的物种有6个,分别为大肠埃希氏菌、肠杆菌科未鉴定属、肠杆菌科、肠杆菌目、变形菌门和γ-变形菌纲,这与Tax4Fun功能预测结果相一致。断奶马驹十二指肠菌群主要为生物体系统(organismal systems)和未分类(unclassified)等功能,空肠菌群主要为代谢(metabolism)等功能,回肠菌群主要为环境信息处理(environmental information processing)等功能。结合本试验测定的空肠和回肠菌群组成,这些碳氢化合物的降解以及潜在致病能力的上升与厚壁菌门和变形菌门的相对丰度是高度相关的,说明空肠是厚壁菌门以及隶属于厚壁菌门等有益菌的定植场所,且饲粮中碳水化合物主要在空肠中进行消化分解,回肠则是大部分隶属于变形菌门的致病菌攻击的主要部位。造成这种差异的原因可能与断奶应激有关,断奶应激能够改变动物肠道菌群组成和数量[27],且导致幼龄动物肠道菌群紊乱,进而使变形菌门、梭杆菌门的多数病原菌属的相对丰度增加[28]。
4 结 论
本试验条件下,断奶马驹十二指肠、空肠和回肠菌群组成存在明显差异,十二指肠和空肠菌群中主要以厚壁菌门及隶属于厚壁菌门的细菌为主,回肠菌群中则主要以厚壁菌门和变形菌门的细菌为主。
