产酸克雷伯菌核心基因组多位点序列分型方案
2023-10-16陈胜林康雨童梁一赫邱小彤刘雪萍李振军
陈胜林,康雨童,梁一赫,徐 帅,邱小彤,李 芳,刘雪萍,李振军
产酸克雷伯菌 (Klebsiellaoxytoca) 是重要的机会性致病菌和医院获得性感染的重要病原体[1-4],严重时可导致抗生素相关性出血性结肠炎[5]。由耐碳青霉烯类肠杆菌科 (carbapenem-resistant Enterobacteriaceae,CRE) 成员产酸克雷伯菌引起的尿路感染对伴有合并症的、免疫功能低下的患者来说可能是致命的[6]。此外,Singh L等[7]的研究显示,产酸克雷伯菌对亚胺培南和美罗培南的耐药率为85%。作为重要的CRE菌株,西班牙的一项研究显示,CRE的产酸克雷伯菌的出现主要是由于产VIM-1和OXA-48的分离株的传播[8],而携带这些高风险的克隆复合群通常是重要的公共卫生威胁[1,5,8]。因此,对产酸克雷伯菌进行高效的基因组分型及识别一些潜在风险的克隆群 (clonal groups,CGs) 非常重要。
序列分型对于评估流行病学关联和确定暴发期间可能的传染源非常重要。目前,主要采用脉冲场凝胶电泳技术 (pulsed-field gel electrophoresis,PFGE)[8-11]和多位点序列分型 (multilocus sequence typing,MLST)[12-13]2种分子分型技术确定产酸克雷伯菌分离株之间的克隆关系。PFGE是医院中广泛使用的一种方法,用于识别产酸克雷伯菌院感暴发,然而这种方法费时、费力,且不容易在不同临床实验室之间重复[14]。MLST的分辨率并不是那么高[15-17]。近年来,由于测序成本大幅降低,基于全基因组测序(whole-genome sequencing,WGS)的方法已经变得可行,并可以在流行病学调查中实现对分离株的高度区分。核心基因组多位点序列分型(core genome multilocus sequence typing,cgMLST)已经对结核分枝杆菌[18]、肺炎克雷伯菌[19]、鲍曼不动杆菌[20]、铜绿假单胞菌[21]等多种病原体进行了高分辨率的分子分型。据我们所知,目前还没有公开可用的产酸克雷伯菌的cgMLST方案。
近年来,随着基因组流行病学的不断发展,作为MLST的延伸,GbG (gene-by-gene) 的分析被提倡[22],这种方法已被全球PulseNet用于利用高通量测序来识别细菌菌株。开源的ChewBBACA算法[23]使一个简单可用的生物信息学管道来创建目标菌株的cgMLST方案变得可行。为了实现产酸克雷伯菌的cgMLST,识别潜在风险的产酸克雷伯菌的CGs,我们使用ChewBBACA作为GbG分析的生物信息学管道,构建并验证了产酸克雷伯菌的cgMLST方案。产酸克雷伯菌复合体是一个由9个物种组成的复合体[24-25],而产酸克雷伯菌是这一复合体中重要的机会致病菌,它是医院获得性感染的主要来源[6-7],并已被证明是引起抗生素相关性出血性结肠炎的致病菌[5]。因此,在这项研究中,我们建立了针对产酸克雷伯菌的cgMLST方案,解析其遗传多样性,为产酸克雷伯菌的监测、防控提供理论依据。此外,我们还将研究不同CGs携带的抗生素耐药基因 (antibiotic resistance genes,ARGs) 和毒力基因,以识别潜在高风险的产酸克雷伯菌的CGs。
1 材料与方法
1.1 来自序列数据库的基因组 从美国国家生物技术信息中心(NCBI)基因组序列库(http://www.ncbi.nlm.nih.gov/genome/)下载了从建库至2022年3月3日期间的461个产酸克雷伯菌(肠杆菌科)的基因组序列。使用GENOME TAXONOMY DATABASE(GTDB)[26]工具对461个基因组进行物种注释,删除了47个非产酸克雷伯菌的基因组序列。丢弃了6个由于基因组组装完成率低于95%或污染率大于5%的基因组序列。
1.2 cgMLST方案的定义 在这项研究中,chewBBACA (https://github.com/B-UMMI/chewBBACA)旨在创建和评估核心基因组GbG的分型方案,然后在产酸克雷伯菌的基因组序列中进行等位基因调用,并且在cgMLST方案创建步骤中所允许的等位基因长度的最小值为201 bp、长度变异在20%以内。
第一部分是cgMLST方案的创建。在研究开始之前,使用产酸克雷伯菌参考基因组FDAARGOS_500 (GenBank序列参考号:GCA_003812925.1) 在 Prodigal v2.6.3[27]中创建一个训练文件,这个文件将在后续的步骤中使用。产酸克雷伯菌 FDAARGOS_500被用作参考基因组仅用于预测全基因组多位点序列分型 (wgMLST) 基因座,不纳入进一步的分析中。第一步共注释21个完整的基因组序列,在此步骤中,chewBBACA算法定义了每个基因组的编码序列(CDs),然后以成对的方式比较基因组中的所有CDs,以生成包含所选 CDs 的单个 FASTA 文件。然后,执行如下两步评估过程以识别基因组中的所有CDs。一是删除与其他CDs具有相同序列但长度较小的CDs,保留较大的CDs; 二是通过执行all-against-all BLASTP搜索并计算爆炸得分比 (Blast Score Ratio,BSR)[28]。BSR 成对比较大于0.6的 CDs 被视为同一基因座(loci)的等位基因,较大的等位基因 (以 bps 为单位) 被保留下来用于定义 cgMLST 方案。从100%可用的完整基因组序列 (21个基因组) 中选择候选基因座来定义我们的cgMLST方案。在这个步骤中,CDs存在的阈值是100%。这种选择最大限度地降低了由于不完整的基因组序列而遗漏核心基因组位点的可能。
第二部分是cgMLST方案的验证。386个不完整的产酸克雷伯菌的基因组序列被用于验证候选基因座。由于验证过程中的使用的是不完整的基因组,我们降低了严格性选择保留99%的产酸克雷伯菌基因组序列共有的候选基因座。
1.3 基于cgMLST方案定义CGs 为了精确地定义CGs,我们分析了成对基因组中等位基因错配(具有不同序列的基因座)数量的分布。与经典 MLST 数据的分类类似,本研究中cgMLST 数据采用单锁链算法对分离株进行准确分类[29]。
1.4 cgMLST方案结果的图形展示 根据cgMLST 方案结果,使用 GrapeTree(2.1 版)为每个产酸克雷伯菌分离株序列获得的等位基因图谱构建最小生成树,并通过 MSTree(2.1 版)方法实现可视化。
1.5 基于cgMLST方案探讨产酸克雷伯菌CGs在国家或地区层面上的遗传多样性 基于cgMLST方案,建立不同国家或地区的最小生成树,并重点关注CGs在不同国家或地区的分布,研究不同国家或地区之间共有的和独特的CGs。
1.6 抗性基因和毒力基因的注释 使用ABRicate 管道 (https://github.com/tseemann/abricate) 比对CARD (https://card.mcmaster.ca/) 数据库和 VFDB ( http://www.mgc.ac.cn/VFs/main.htm) 数据库注释抗性基因和毒力基因。抗性基因和毒力基因的注释参数为基因覆盖率大于95% 和同一性大于85%。
2 结 果
2.1 纳入的产酸克雷伯菌基因组序列特征 除1个产酸克雷伯菌参考基因组被用于预测wgMLST基因座外,407株产酸克雷伯菌基因组序列被用于执行产酸克雷伯菌的cgMLST方案。根据我们的cgMLST方案,400个产酸克雷伯菌的基因组序列被纳入最终的分型分析。400株产酸克雷伯菌分离株至少分布在18个国家或地区,并且绝大多数分离菌株收集于患者(89.8%)。
2.2 产酸克雷伯菌的cgMLST方案 在cgMLST方案创建步骤中,以FDAARGOS_500为参考基因组,使用21个完整的产酸克雷伯的基因组序列,共注释了9 735个目标基因座,生成了全基因组多位点序列分型(wgMLST)数据集(图1)。在进一步的过滤步骤中,使用 RemoveGenes 操作从 wgMLST 方案中删除了67个旁系同源基因座,TestGenomeQuality (基因组质量测试)操作(阈值 65)过滤掉额外的 5 583个基因座。至此,总共保留了4 085个cgMLST 候选基因座,这些候选基因座存在于 20 个完整的产酸克雷伯菌基因组序列中。在此步骤中,1个完整的基因组序列P620(GenBank序列参考号:GCA_009707385.1)被丢弃,该菌株序列在基因组质量测试(阈值为65)操作中被移除,因而不包含4 085个cgMLST候选基因座。
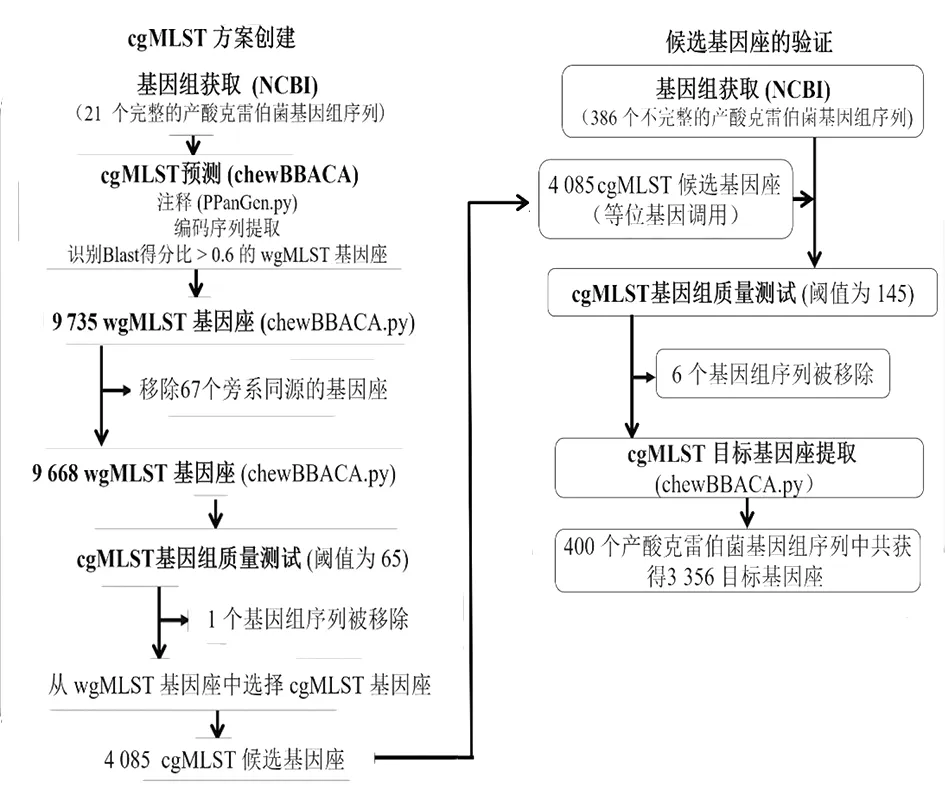
图1 使用chewBBACA开发的产酸克雷伯菌的核心基因多位点序列分型 (cgMLST) 方案的详细流程图
在cgMLST方案验证步骤中,386 个不完整的产酸克雷伯菌基因组序列被用来验证 4 085个cgMLST候选基因座。使用Test Genome Quality (基因组质量测试)操作(阈值 145)移除了 6 个不完整的产酸克雷伯菌的基因组序列(GenBank序列参考号:GCA_001063775.1、GCA_001065715.1、GCA_002414045.1、GCA_900451235.1、GCA_900451255.1、GCA_900478285.1)。最终,在验证步骤后,共3 356个cgMLST目标基因座被保留。据此,定义了一个由3 356个核心基因组成的cgMLST方案(以下统称为3356-cgMLST方案),涵盖了参考基因组FDAARGOS_500预测的5 378个开放阅读框中的62.4%。
2.3 基于3356-cgMLST方案定义产酸克雷伯菌的CGs 使用单连锁算法[29]对3 356 目标核心基因进行精确聚类。几乎没有成对的菌株之间等位基因错配数分布在221~1 230(图2)。因此,根据成对等位基因错配数量的分布(即某对菌株序列的基因座数量差异)的结果,我们选择 221 个等位基因的差异作为阈值作为定义CGs的临界值。将基于3356-cgMLST方案的产酸克雷伯菌的CGs定义为一组3356-cgMLST 配置文件,该配置文件与该组的至少一个其他成员相差不超过 221个等位基因错配。根据该定义,最终识别出130个产酸克雷伯菌的CGs。
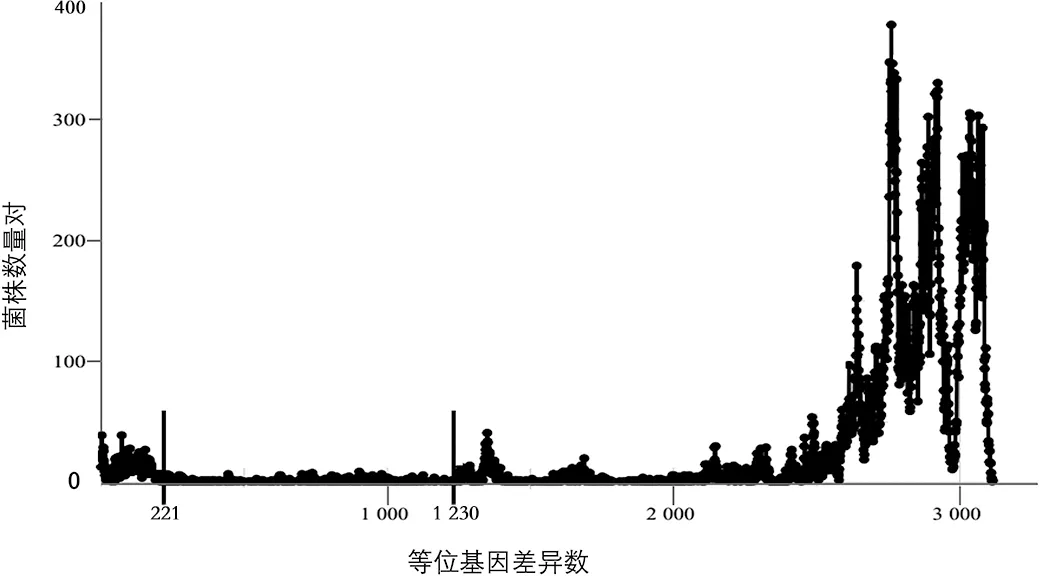
图2 产酸克雷伯菌中成对等位基因错配数量(等位基因差异数)的分布情况
2.4 3356-cgMLST方案结果的可视化—最小生成树的构建 根据3356-cgMLST方案,400株产酸克雷伯菌基因组序列共获得 130 种CGs。由于最小生成树可视化的限制,图 3 仅展示了携带菌株数量≥3的CGs。从图3可以看出,CGs呈现出良好的成簇分布现象。也就是说,根据所定义的 3356-cgMLST方案,绝大多数产酸克雷伯菌基因组序列均得到了很好的分型。

图3 基于cgMLST的400株产酸克雷伯菌分离株克隆群(CGs)的最小生成树(MST)
2.5 基于3356-cgMLST方案探讨产酸克雷伯菌CGs在国家或地区层面上的遗传多样性 基于3356-cgMLST方案,400个产酸克雷伯菌基因组序列的最小生成树显示,产酸克雷伯菌呈现出明显的多国家多地区传播现象(图4)。表1给出了18个国家或地区的CGs分布情况。38个共有的CGs分布在13个国家或地区。92个独特的CGs分布在14个国家或地区。CG5分布在9个国家或地区,CG26分布在6个国家或地区,CG15和CG29分布在5个国家或地区。澳大利亚有23个独特的CGs,美国有21个独特的CGs,英国有12个独特的CGs。总的来说,产酸克雷伯菌CGs呈现出明显的遗传多样性。

表1 产酸克雷伯菌的克隆群在不同国家或地区中的分布
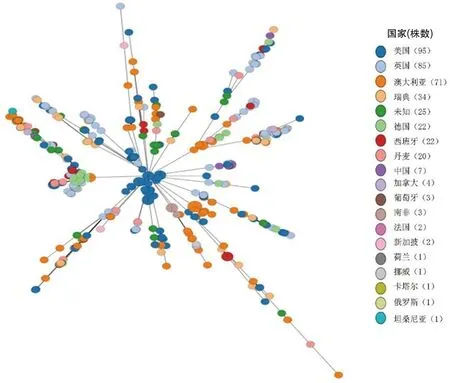
图4 不同国家或地区分离的400株产酸克雷伯菌cgMLST谱系的最小生成树
2.6 抗性基因和毒力基因在产酸克雷伯菌CGs中的分布 产酸克雷伯菌CGs中与抗生素耐药有关的基因。在400株产酸克雷伯菌分离株中发现了136种ARG的亚型。产酸克雷伯菌CGs中CRP、H-NS、Klebsiella_pneumoniae_KpnF、Klebsiella_pneumoniae_KpnG、LptD、OmpA和oqxB等ARG携带率超过95%。不同CGs携带ARG亚型的数量为7~61种。IMP-1仅存在于CG1中。Ode T等[30]对多重耐药的产酸克雷伯菌的耐药机制分析结果表明,IMP-1金属β-内酰胺酶 (metallo-β-lactamase,MBL),这个产酸克雷伯菌产生的约140 kb的质粒介导了其对β-内酰胺类、喹诺酮类和氨基糖苷类的耐药性转移。重要的产CRE基因NDM-7仅存在CG27中。而OXA-181只存在于CG118中。OXA-181是OXA-48的近亲[31],与OXA-48只有4个aa的区别[32],是重要的产CRE基因。这些CGs可能是潜在高风险的产酸克雷伯菌的CGs。
产酸克雷伯菌CGs中与毒力有关的基因。在400个产酸克雷伯菌基因组序列中共发现了35种毒力基因。毒力基因AllA、AllC、AllD、AllS、entB和fepC在130中种CGs中的携带率为100%。毒力基因acrB、allB、allR、fyuA、gnd、irp1、irp2、mrkA、rcsB、ybdA、ybtA、ybtP、ybtQ和ybtS的携带率高于95%。不同CGs携带毒力基因的种类数量没有明显差异。但值得注意的是,毒力基因astA是产酸克雷伯菌的一个蛋白编码基因,编码精氨酸N-琥珀酰转移酶,仅存在CG111中;毒力基因hcp/tssD主要编码假定蛋白,仅存在CG68中;毒力基因tcpC主要编码结合转移蛋白,仅存在CG27中。6种独特的毒力类型 (kpsC、kpsD、kpsE、kpsF、kpsS、kpsU) 仅在CG12中出现,并且主要编码荚膜多糖修饰蛋白,是产酸克雷伯菌致病性的重要来源。这些CGs可能是潜在的高风险的产酸克雷伯菌的CGs。
3 讨 论
产酸克雷伯菌是引起医院获得性感染的重要病原体[1-4]。产毒素的产酸克雷伯菌引起的抗生素相关出血性结肠炎是一个重要的公共卫生问题[33-36]。因此,一个高分辨率的产酸克雷伯菌的分型方案非常重要。目前,PFGE[9,36-37]和MLST[38-39]这2种分型方法的主要缺点是分辨率低[15-17]和难以实现PFGE的标准化[14]在实验室间的比较。全球细菌监测计划和医院环境卫生中的疫情调查需要简单、快速和准确的分型方法。基于全基因组测序(WGS)的cgMLST分型已被证明是细菌分型、疾病暴发调查、来源追踪和细菌病原体监测的有效工具[40-42]。cgMLST实现了不需要特定暴发的参考,从整个物种样本中确定合适的、无偏倚的可能集群的平均值[43]。我们使用开源的ChewBBACA算法[23]创建和验证了首个产酸克雷伯菌的cgMLST方案。
本研究中,我们基于20个完整的基因组序列和380个不完整的基因组序列共获得了3 356个核心基因。在这个过程中,共有1个完整的基因组序列和6个不完整的基因组序列被移除。在创建cgMLST方案的步骤中移除1个完整的基因组序列,是因为需要对基因组序列中的核心基因进行严格限制,即目标基因座在基因组序列中100%存在,以便最大程度地提高方案的准确性。而在cgMLST方案的验证步骤中6个不完整的基因组序列被移除,是因为这6个不完整的基因组序列无法保证候选基因座在其基因组序列中99%存在。这7个被移除的基因组序列可能与纳入分型的其他400个基因组序列相比有较大的基因组变异。总的来说,3356-cgMLST方案对本研究中纳入的超过98%(400/407)的产酸克雷伯菌的基因组序列成功地进行了分型。提出的由3 356个核心基因构成的 cgMLST方案将400个产酸克雷伯基因组序列划分为130个不同的CGs,CG26 (11.0%)是最主要的优势CG,其次是CG1 (9.3%)、CG5 (8.3%)、CG15 (5.0%) 和CG8 (3.3%)。基于3356-cgMLST方案获得的CGs分布情况可以看出产酸克雷伯菌呈现出明显的多国家多地区传播现象,这也在一定程度上提示了产酸克雷伯菌基因组序列呈现出高度的遗传多样性。我们在GitHub (https://github.com/Natasha22222222/cgMLST-Klebsiella-oxytoca) 上提供了关于3356-cgMLST方案创建及验证步骤中的所使用的基因组序列、详细的cgMLST过程文件以及3 356个核心基因构成,以促进更深一步的讨论并希望能够有助于建立产酸克雷伯菌的cgMLST共识。
ARG亚型和毒力基因的分布似乎与CGs有关。毒力基因astA编码一种热稳定的肠毒素,只在CG111中出现。kpsC、kpsD、kpsE、kpsF、kpsS和kpsU只存在于CG12中,这些毒力基因与运输蛋白KpsFEDUCS(KpsF、KpsE、KpsD、KpsU、KpsC和KpsS)[44-45]和重要荚膜分子的编码有关。我们仅在CG118中发现了OXA-181,这是不同地理区域最常见的OXA-48样变体之一[32]。OXA-181阳性的肺炎克雷伯菌已成为重要的CRE生产者,先后出现在荷兰和新西兰等地[31,46-47],成为一个严重的公共卫生问题。德国的一项研究表明可移动遗传元件ColKP3质粒的存在可介导OXA-181的耐药转移[48]。这些都提示携带OXA-181的CG118是潜在的高风险的产酸克雷伯菌的CG。此外,我们还在CG27中发现了重要的产CRE基因NDM-7[49]。CRE基因的水平传播,由携带额外抗性元件的可移动遗传元件介导,赋予对各类抗生素的抗性,导致多重耐药性[50-51]。多重耐药的克雷伯氏菌感染的暴发给住院病人带来重大的生命健康威胁[36,52-53]。特别是最近来自意大利的一项研究表明,CRE的肺炎克雷伯菌中抗生素耐药基因和毒力因子的共存导致肺炎克雷伯菌新克隆群的出现[54],并可能对解决抗菌素耐药性构成重大挑战。因此,在产酸克雷伯菌中观察到的这种潜在的高风险的CGs应该给予高度重视。
综上所述,我们利用开放的平台chewBBACA结合细菌基因组测序,提出了产酸克雷伯菌的首个cgMLST方案。我们的目的是促进讨论并希望可以有助于建立一个关于产酸克雷伯菌的cgMLST共识。我们将基于3356-cgMLST方案精确定义的CGs与注释到的抗性基因和毒力基因相结合,发现了产酸克雷伯菌的潜在的高风险CGs。但由于本研究所涉及的国家或地区及其菌株数量有限,cgMLST方案仍需要更多数量及来源的产酸克雷伯菌基因组序列进一步验证。
利益冲突:无
引用本文格式:陈胜林,康雨童,梁一赫,等.产酸克雷伯菌核心基因组多位点序列分型方案[J].中国人兽共患病学报,2023,39(9):912-919,926.DOI:10.3969/j.issn.1002-2694.2023.00.095
