疱疹病毒引起内质网应激/未折叠蛋白反应研究进展
2023-10-15蔡海情王微曾茂芹毕文文陈莉张黔东袁阳文明
蔡海情, 王微, 曾茂芹, 毕文文, 陈莉, 张黔东, 袁阳, 文明,2*
(1.贵州大学动物科学学院,贵阳 550025; 2.贵州省动物生物制品工程技术研究中心,贵阳 550025)
疱疹病毒(Herpesvirus)是一类有囊膜的双链DNA 病毒,其基因组大小约125~241 kbp,包含70~170 个基因,结构复杂,由对称的正二十面体结构和非对称成分组成。一般来说,疱疹病毒与宿主共同进化并高度适应宿主,宿主包括许多哺乳动物、鸟类、爬行动物和人等[1]。初次感染后他们能够建立终身潜伏感染,在此期间病毒反复感染,给人和动物带来了重大损害。其中,人类Ⅰ型单纯疱疹病毒(herpes simplex virus-Ⅰ, HSV-Ⅰ)可引起人体蔓延性皮炎,在宿主细胞中呈潜伏感染且终生带毒,严重危害人类健康[2];马立克病病毒(marek’s disease virus,MDV)、传染性喉气管炎病毒(infectious laryngotracheitis virus,IBDV)、鸭肠炎病毒(duck enteritis virus,DEV)和伪狂犬病病毒(pseudorabies virus,PRV)等引起畜禽的多种器官损伤和免疫性疾病,严重威胁畜禽养殖业健康发展,造成了严重的经济损失。
通常情况下,疱疹病毒复制涉及病毒粒子被膜蛋白与细胞表面受体相互作用,然后通过细胞表面的膜融合或内吞作用进入,脱壳并被运送到核孔中,基因组进入细胞核[3]。病毒早期基因编码DNA 复制并合成多种蛋白质,参与调节宿主细胞新陈代谢或免疫反应,晚期基因主要编码病毒粒子蛋白。由于疱疹病毒复制给宿主内质网(endoplasmic reticulum,ER)带来了过重的负担,以产生病毒蛋白并促进病毒颗粒运输,从而引起内质网应激(endoplasmic reticulum stress,ERS)。为了应对ERS 和维持ER 蛋白平衡,细胞启动未折叠蛋白反应(unfolded protein response,UPR)[4]。如单纯疱疹病毒(herpes simplex virus,HSV)必需借用宿主细胞的蛋白合成系统合成自身蛋白,用于组装子代病毒粒子;细胞出于自我保护会启动ERS 介导的蛋白降解、炎症反应、自噬、凋亡等抑制HSV 病毒蛋白的合成、降解病毒蛋白或诱导伴侣分子表达,从而诱导UPR[5];MDV 在复制时诱发严重的免疫抑制,导致促炎细胞因子过度表达,激活UPR 可能单独或与其他先天传感途径协同作用,导致核转录因子κB(nuclear factor-kappa B,NF-κB)被激活,介导免疫病理学和神经损伤[6]。同HSV 和MDV 一样,DEV 和PRV 也会引发ERS,激活UPR,导致炎症反应的加剧和其他级联反应[7-8]。
内质网是合成分泌蛋白和膜蛋白、蛋白翻译后修饰、调控钙离子储存和脂类合成的细胞器,具有重要生理功能[9]。一般情况下,新生蛋白在分子伴侣和折叠酶(统称为内质网伴侣)的作用下折叠,正确折叠的蛋白质被运输到高尔基体进一步加工,形成分泌蛋白,进入内膜系统或被分泌到细胞外发挥作用。但是,当细胞生长条件发生变化(如DNA 损伤、化学诱导、微生物感染等)时,内质网蛋白质发生未折叠或错误折叠,未折叠或错误折叠的蛋白质就会被保留在内质网中,通过内质网相关降解机制(endoplasmic reticulum associated degradation,ERAD)反转运到细胞质中,由蛋白酶体降解。一般情况下上述蛋白会被激活的ER 分子伴侣和ERAD 处理,当细胞合成的分泌蛋白质超过折叠装置和ERAD 的能力时,未折叠的蛋白质就会大量堆积在内质网中,并倾向于形成蛋白质聚集体,进而导致细胞蛋白质合成功能的异常甚至引起细胞凋亡。为了缓解这种应激状态,真核细胞激活了一系列的自我防御机制,统称为未折叠蛋白反应(UPR),从而恢复内质网稳态[10-14](图1)。近年来,国内外研究人员对疱疹病毒的研究逐渐增多,主要集中在HSV[15]、PRV[16]、MDV[17]和DEV[18]等,本文概述了上述疱疹病毒对ERS 及UPR 之间的相互作用,以期为研究其相关分子机制提供参考。
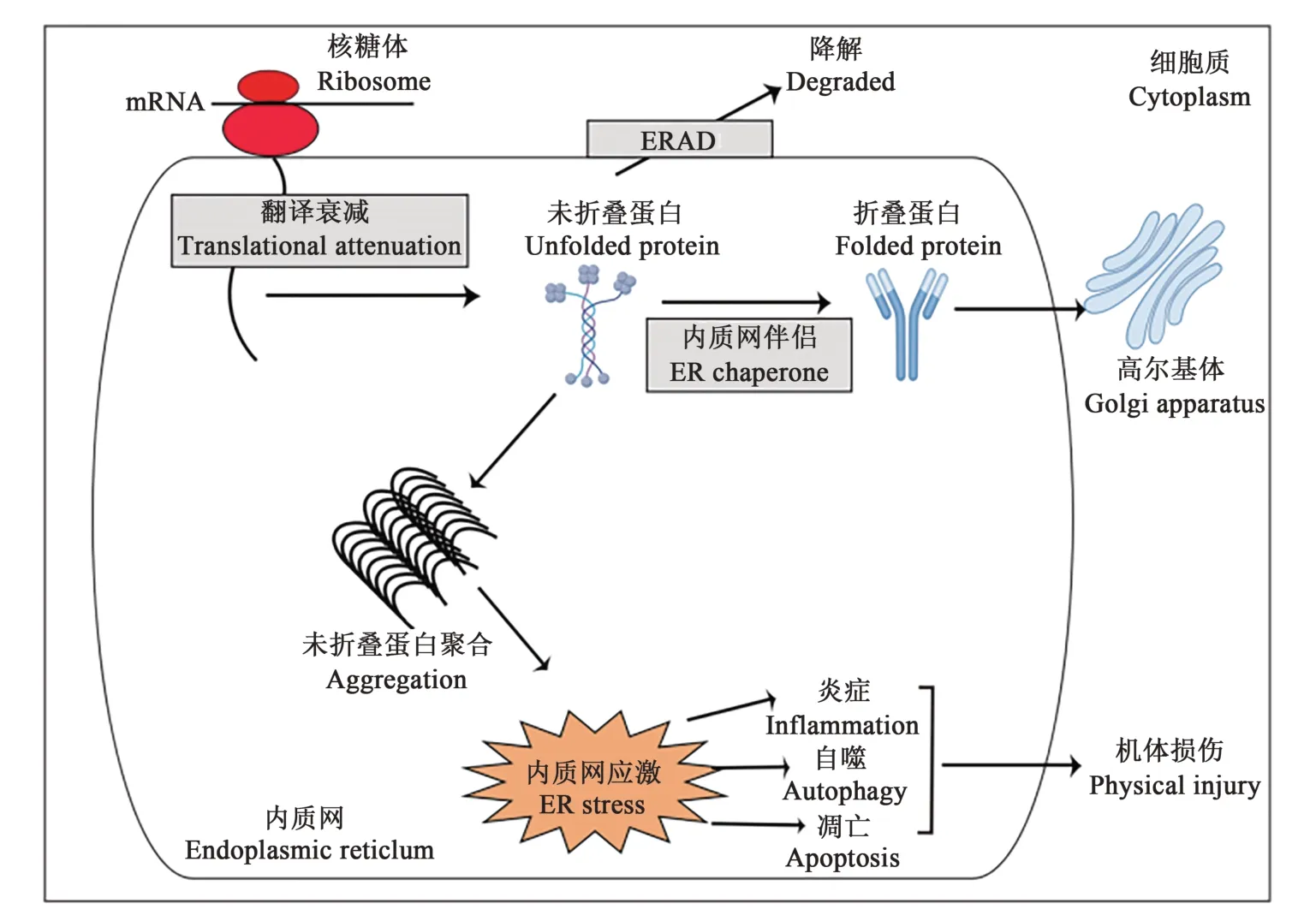
图1 内质网应激反应Fig. 1 Endoplasmic reticulum stress response
1 内质网应激/未折叠蛋白对病毒的反应机制
在病毒感染后,细胞激活未折叠蛋白反应通路,使它们免受内质网应激(ERS)所诱导的凋亡反应[19]。病毒复制时,其劫持宿主翻译装置,产生大量蛋白,内质网的动态平衡遭到破坏,导致错误折叠和未折叠蛋白的快速积累,激活ERS,进而启动未折叠蛋白反应(UPR)[20]。一些病毒甚至利用内质网作为复制位点,以达到在细胞内快速增殖的目的,如丙型肝炎病毒(hepatitis C virus,HCV)[21]和非洲猪瘟病毒(African swine fever virus,ASFV)[22]。此外,大量研究发现乙型肝炎病毒(hepatitis B virus,HBV)[23]、黄病毒(flavivirus)[24]、博尔纳病毒(borna virus)[25]等均可引起ERS,机体为减轻ERS 利用UPR 来恢复内质网的相对稳定。哺乳动物细胞中,病毒引起的ERS主要由以下ER跨膜受体介导:RNA 样蛋白激酶样内质网激酶(protein kinase RNA-like ER kinase,PERK)、肌醇需求激酶1(inositol-requiring kinase 1,IRE1)及激活转录因子6(activating transcription factor 6,ATF6)。在正常细胞中3 个感受器与内质网驻留伴侣免疫结合球蛋白(binding immunoglobulin protein,BIP)或葡萄糖调节蛋白78(glucoseregulated protein 78,GRP78)结合而维持抑制状态,当细胞发生ERS 时,BIP 优先与错误折叠蛋白质结合,PERK 和ATF6 解离以减轻应激[26-27]。PERK 使真核细胞起始因子2 的α 亚基(α-subunit of eukaryotic initiation factor 2,eIF2α)磷酸化,终止鸟嘌呤三核苷酸磷酸(guanosine triphosphate,GTP)与鸟嘌呤二核苷酸磷酸(guanosine diphosphate,GDP)的交换作用,从而减缓或暂停蛋白质合成,缓解内质网的超负荷。ATF6则上调ER 蛋白和ERAD 表达,扩大内质网折叠能力来缓解ERS。IRE1 被未折叠蛋白的直接结合激活,并剪接XBP1(X box-binding protein 1),XBP1 是活跃的转录因子,通过上调多种折叠酶及与蛋白质折叠相关的UPR目标基因,参与蛋白质转运、脂质合成等过程以校正内质网稳态[28]。活化的IRE1、ATF6 和PERK 可分别通过各自的信号通路启动UPR。
1.1 PERK-eIF2α通路
PERK 属于丝氨酸/苏氨酸蛋白激酶,与IRE1同为Ⅰ型跨膜蛋白并且结构相似,ER腔中感测到错误折叠或未折叠蛋白质时,PERK 与BIP/GRP78发生解离,形成二聚体,引发反式自磷酸化,激活下游信号通路[29]。PERK 经寡聚和自动磷酸化形成活性PERK 使真核细胞翻译起始因子2 的α 亚基(eIF2α)磷酸化,减少内质网蛋白质合成,缓解ERS[30-31]。然而,这种数量有限的活性eIF2α 会选择性地增加激活转录因子4(activating transcription factor 4,ATF4)的翻译。ATF4 是一种促进细胞生存的转录因子,激活后可参与细胞凋亡、生物合成和胞内氧化还原反应等过程[32]。ATF4 诱导生长阻滞和DNA 损伤诱导蛋白34(growth arrest and DNA damage-inducible protein34,GADD34),它是蛋白磷酸酶1(PP1)的调节亚基,可引起生长停滞和诱导DNA 损伤,其招募PP1 磷酸化eIF2α,使PERK-eIF2α 下调,减轻翻译衰减,最后在UPR中构成1 个负反馈环[33],维持蛋白质合成过程中的“起始-终止”这一动态平衡。但在过度、持久的内质网应激条件下,活化的PERK 无法维持这一动态平衡,细胞无法恢复到正常的生理状态,就会启动调控细胞凋亡的未折叠蛋白反应信号通路PERK-ATF4-CHOP。凋亡相关蛋白CHOP(pro-apoptotic CCAAT/enhancer-binding protein homologous protein)是一种29 kD 的蛋白质,具有169 个(人类)或168 个(啮齿动物)氨基酸残基[34],属于CCAAT/增强子结合蛋白家族,参与编码、增殖、分化、表达以及能量代谢等基因的调节。研究证明CHOP 包含2 个功能结构域,1 个N 端转录激活结构域和1 个C 端碱性亮氨酸拉链(basicleucine zipper,bZIP)结构域,bZIP 在诱导细胞凋亡中起着至关重要的作用[35-36]。CHOP 介导的GADD34 激活促进elF2α 蛋白去磷酸化,逆转翻译抑制。内质网释放翻译有助于未折叠蛋白的积累,同时允许编码促凋亡蛋白的mRNAs 翻译[37]。因此,CHOP被认为是ERS诱导凋亡蛋白最重要的介导物之一,参与多种基因的调控及表达[38],如CHOP、生长阻滞和DNA 损伤诱导蛋白153(growth arrest and DNA damage-inducible protein153,GADD153)在UPR 中发挥协同作用,激活凋亡相关靶点GADD34、死亡受体5(death receptor-5,DR5)、内质网氧化还原酶-1(endoplasmic reticulum oxidoreductase 1,ERO1)等促进细胞凋亡[37]。
1.2 IRE1-XBP1通路
IRE1是含有丝氨酸-苏氨酸激酶结构域和内切核酸酶结构域的双活性酶[39]。IRE1 在哺乳动物中存在2 种异构体—IRE1α 和IRE1β,IRE1α 主要存在于哺乳动物细胞中,IRE1β 一般在消化道上皮细胞中表达[40]。当IRE1从GRP78解离时,通过同源二聚化和反式自磷酸化机制促进IRE1α的活化,进而激活苏氨酸激酶和核酸内切酶活性。活化的IRE1 从XBP1 中移除26 个核苷酸组成的内含子,形成剪接的XBP1(S),随后被翻译成bZIP 转录因子,进而激活增强ER 蛋白折叠能力、磷脂生物合成和ERAD 基因的转录[41-42]。XBP1(S)还能激活Hsp40 家族成员P58IPK,P58IPK 可以结合PERK 并抑制其对磷酸化eIF2a 的活性。IRE1 本身可以通过调节其依赖衰变(regulated IRE1-dependent mRNA decay,RIDD)途径降解ER结合的mRNAs,从而限制蛋白质翻译并降低ER管腔内部蛋白质折叠负荷[43]。但在内质网持续应激时,胞质中游离的IRE1 与肿瘤坏死因子α 受体相关因子2(tumor necrosis factor receptor associated factor 2,TRAF2)结合并使其活化,TRAF2 能进一步激活NF-κB 和C-jun N 末端激酶(c-jun-Nterminal kinase,JNK)通路,从而激活细胞内的炎症反应诱导细胞凋亡[44]。在ERS 的早期阶段,JNK 介导的B 细胞瘤蛋白2(B-cell lymphoma protein-2,BCL-2)磷酸化导致Beclin1 的解离和自噬激活[45]。此外,IRE1-XBP1 信号通路在病毒感染中起着重要作用。首先,病毒感染后ERAD 的激活加速了蛋白质的降解[46];其次,CHOP 诱导凋亡,并在诱导自噬中发挥作用;第三,IRE1 介导内质网中部分mRNA 选择性降解,减少宿主相关蛋白产生,促进自身复制,但每种病毒由IRE1-XBP1介导的细胞反应不同[47]。
1.3 ATF6途径
ATF6 是一种bZIP 转录因子,但最初是作为Ⅱ型跨膜蛋白合成的,其含有内质网应激敏感的管腔结构域、跨膜结构域和胞质N-末端结构域[27]。ATF6作为ERS的下游启动因子,由内质网衍生的囊泡包装,并移位到高尔基复合体,在高尔基复合体中被丝氨酸蛋白酶位点1(serine protease site-1,S1P)切割管腔结构域,N-端部分随后被金属蛋白酶位点2(metalloprotease site-2 protease,S2P)切割,释放含活性的N 端DNA 结合域ATF6(N)[48]。ATF6(N)移位到细胞核并激活提高ER 折叠能力的基因,如ERS 反应元件(endoplasmic reticulum stress element,ERSE)基因、BIP、GRP94、蛋白二硫化物异构酶(protein disulfide isomerase,PDI)、CHOP 等,同时激活XBP1 和ERAD 等基因的转录[49]。
总体而言,这3 个分支通过减少内质网多肽链的合成、降解内质网定位的蛋白和扩大内质网折叠能力来修复内质网应激(图2)。

图2 未折叠蛋白反应通路Fig. 2 Unfolded protein reaction pathway
2 疱疹病毒与内质网应激
根据国际病毒分类委员会(International Committee on Taxonomy of Viruses,ICTV)的定义,疱疹病毒包含ɑ、β、γ 3 个亚科,还有其他新发现未分类的,如鹦鹉酸ɑ 疱疹病毒5、猕猴γ 疱疹病毒1等[1]。疱疹病毒在自然宿主范围内受到限制,并高度适应宿主,严重感染通常只在胎儿、免疫功能低下的人或某些宿主中体现。疱疹病毒作为胞内寄生生物,在细胞内的复制有一套自己的控制系统,在其感染期间,病毒能够劫持宿主的翻译机器,大量合成自身相关蛋白,使内质网充满病毒蛋白,导致内质网直接或间接发生应激,引起未折叠蛋白反应[50]。病毒蛋白在内质网中的积聚对蛋白质折叠提出了更高的要求,可能导致内质网稳态失衡甚至影响宿主细胞的存活。疱疹病毒不仅利用宿主细胞器来生产病毒糖蛋白,甚至利用内质网作为复制位点来组装子代病毒。以下集中概述HSV-Ⅰ、PRV、MDV 和DEV 等疱疹病毒对ERS 及UPR之间的的相互作用,
2.1 Ⅰ型单纯疱疹病毒
Ⅰ型单纯疱疹病毒(HSV-Ⅰ)属于α疱疹病毒亚科,感染细胞后引起ERS,通过调控UPR 和ERAD 机制促进病毒复制,抑制细胞凋亡。Burnett 等[51]研究发现,HSV-Ⅰ在感染早期可有效解除UPR,只激活ATF6 信号通路,而PERK 和IRE1/XBP1 通路被抑制;在HSV-Ⅰ感染后期,eIF2α/ATF4 信号臂活性增加,提示病毒粒子组装和输出完成,释放UPR 信号转导的抑制;同时发现,插入病毒序列的ICP0 真核载体启动子对ERS有明显的响应,提示HSV-Ⅰ可能利用ICP0作为传感器来调节细胞的应激反应。Zhang 等[52]研究HSV-1 UL41 蛋白在抑制IRE1/XBP1 时发现,UL41 的异位表达使XBP1 翻译降低,并阻断ERS诱导剂毒胡萝卜素(thapsigargin,Tg)诱导的XBP1剪接激活;野生型HSV-1(wild type HSV-1,WT-HSV-1)可降低Tg 诱导的XBP1mRNA 转录,而UL41缺失突变体R2621 则不能;然而,未经药物预处理的WT HSV-Ⅰ和R2621 均可降低XBP1(S)的mRNA 转录和蛋白翻译,推测在R2621感染过程中可能存在其他机制对XBP1(S)有抑制作用;同时发现HSV-Ⅰ可以通过抑制IRE1/XBP1 来避免可能对病毒复制不利的细胞反应,这可能与XBP1(S)靶基因编码的ERAD 蛋白降解病毒蛋白有关。此前XBP1(S)也被报道能增强树突状细胞中β 干扰素的产生,从而抑制该病毒的复制[53]。因此HSV-Ⅰ感染细胞后发生UPR 的分子机制还需要进一步深入研究。
2.2 伪狂犬病病毒
伪狂犬病病毒(PRV)属于α 疱疹病毒亚科,通过破坏内质网的动态平衡,诱导ERS 并触发未折叠蛋白反应。Yang 等[54]研究发现,PRV 感染的早期阶段ERS 标志物GRP78表达增加,提示ERS 被激活,而随着时间的延长,GRP78的表达不再增加,表明PRV 只有在感染早期诱导ERS反应,且应激反应强弱与病毒复制水平并无平行关系,且在感染24 h时达到峰值;同时对UPR 3个分支的检测发现,IRE1-XBP1 和eIF2α-ATF4 通路被激活,并通过PERK-eIF2α-CHOP-Bcl2 轴诱导细胞凋亡;在整体水平检测ERS 与病毒复制的关系时用毒胡萝卜素(Tg)、牛磺熊去氧胆酸(tauroursodeoxycholic acid,TUDCA)和衣霉素(tunicamycin,Tm)处理细胞,其中Tg 处理过的细胞能使GRP78过量表达,显著上调PRV 的复制,TUDCA 和Tm 下调病毒复制。Chen 等[16]在研究猪圆环病毒二型和PRV 合并感染时发现,ERS 可以通过PERK-eIF2α-ATF4-CHOP 途径激活,且在感染36~72 h 内PRV 抑制猪圆环病毒二型的增殖;进一步的研究表明,PRV 在二者合并感染中起主导作用,并进一步激活IRE1-XBP1-EDEM 途径,一定程度上增强了细胞凋亡。但关于PRV 激活ERS 引起UPR、炎症反应、细胞自噬、凋亡等生物学过程之间的级联机制尚不明确。
2.3 马立克病病毒
马立克病病毒(MDV)属于疱疹病毒科、α 疱疹病毒亚科、马立克病病毒属。Neerukonda 等[17]在研究马立克病病毒感染诱导UPR 时发现,MDV激活UPR,通过增加GRP78/BIP 表达、促进XBP1剪接和诱导α-甘露糖苷酶样蛋白表达等增强ERS;MDV 在体内感染过程中可检测到部分UPR激活,其调节水平受MDV 癌蛋白meq 的影响,其后在MDV 诱导的原发性淋巴瘤中发现ATF6被激活,提示ATF6在肿瘤发展中起一定作用[55]。MDV编码的极早期或早期蛋白可能会阻断UPR 激活,防止UPR 感受器长时间激活而导致的细胞凋亡,以促进自身复制。
2.4 鸭肠炎病毒
鸭肠炎病毒(DEV)又称鸭瘟病毒(duck plague virus,DPV),是疱疹病毒科α 疱疹病毒亚科的成员。DEV 感染会引起雏鸭的组织损伤、淋巴管受损和血管损伤等,是一种急性、败血症传染病[56]。Yin 等[57]发现DEV 感染触发机体ERS,表现为GRP78 表达增加和内质网形态的扩张,DEV 复制增加;感染鸭胚成纤维细胞后,PERK和IRE1被激活,二者还可能参与DEV 诱导的自噬。Zhang等[18]利用中国标准攻毒株DEV(DEV-CSC)诱导鸭胚成纤维细胞,DEV-CSC 感染可显著降低DEF 细胞内Ca2+水平,抑制细胞活力,诱导细胞凋亡,提示DEV-CSC 感染显著上调CHOP、GRP78 和ATF6的表达,可能是由于ERS 形成的微环境有利于DEV 的复制,但其中的分子机制和相关联级反应还有待研究,特别是促凋亡机制;大剂量DEVCSC 感染通过Ca2+介导的ERS/UPR 激活细胞凋亡,这一机制可能与钙网蛋白有关。在DEV 感染后,钙网蛋白在Ca2+稳态和ERS/UPR 诱导中的作用也需要进一步的研究。
2.5 其他疱疹病毒
除了上述几种疱疹病毒外,还有其他多种疱疹病毒及其亚型引起内质网的应激并激活未折叠蛋白反应。如γ疱疹病毒亚科的卡波西肉瘤相关疱疹病毒(kaposis sarcomaassociated herpesvirus,KSHV)[58-59]、疱疹病毒家族β亚群的原型致病成员之一人巨细胞病病毒(human cytomegalo virus,HCMV)[60-61]、水痘带状疱疹病病毒(varicellazoster virus,VZV)[62]、Epstein-Barr 病毒(Epstein-Barr virus,EBV)[63]等,均可不同程度地激活未折叠蛋白的1 条或几条信号通路,从而抑制或促进病毒的复制,影响细胞的凋亡。
3 展望
疱疹病毒是无处不在的双链DNA 包膜病毒,会造成终生感染并导致一系列疾病。其不同成员在感染过程中激活的ERS 所表现出的对病毒感染、复制的影响既有共性,又呈现出差异。病毒诱导的差异可能与糖基化、磷酸化蛋白数量和位置等有关,也与病毒复制时宿主细胞与特定病毒受体结合有关,被不同的伴侣分子识别,从而进入不同的加工成熟路径,并由此向内质网压力感受系统传导不同水平的压力信号。此外,ERS 反应是保守的应激反应,可以感知和响应内质网蛋白稳态,UPR 作为对ERS 的响应,通过转录因子PERK、IRE1 和ATF6 的联合作用,减弱蛋白质瞬时翻译,选择性地上调反应基因,提高蛋白质折叠能力,并降解末端错误折叠的蛋白质,使内质网恢复稳态。但是,由于疱疹病毒基因组编码多达11种不同的囊膜糖蛋白,其复制和转录翻译必然占用大量内质网资源,极易超出内质网稳态,造成ERS 和UPR,最终介导细胞的凋亡。部分疱疹病毒似乎已经进化出某种篡改未折叠蛋白反应的机制,很可能抑制宿主细胞抗病毒转录反应,或促进自身复制的相关基因转录。到目前为止,有效的ERS 诱导药物毒胡萝卜素和衣霉素在UPR 研究中发挥了很大作用,但这些药物通常不能概括病理状态下的ERS,而且它们也有已知的靶点效应。所以研究疱疹病毒自然感染引起ERS 之间的分子机制和病毒与宿主之间反馈机制,可发现病毒复制、转录和翻译的关键节点,为疫苗研制和抗病毒药物研究奠定理论基础。但这些途径仍有许多未知之处,但越来越清楚的是,ERS 和UPR 不仅关联疱疹病毒还与许多人类疾病密切相关。探究UPR 的分子机制对许多人类疾病和动物疫病以及设计通过调节UPR 来治疗疾病或疫病具有重要意义。
