亚胺还原酶的研究进展
2023-10-10朱鑫鑫朱振宇王红月王盼琳沈昕元崔云松陈飞飞许建和郑高伟
朱鑫鑫,朱振宇,王红月,石 敏,王盼琳,沈昕元,崔云松,金 添,陈飞飞,许建和,郑高伟
(华东理工大学生物反应器工程国家重点实验室,上海 200237)
手性胺广泛存在于各种药物、天然产物和精细化学品中(图1)。美国食品药品监督管理局(FDA)批准上市的小分子药物中约有40%含有手性胺的结构砌块。鉴于酶催化剂在不对称合成中近乎完美的立体选择性。故而亟待开发出一种高效绿色的酶催化剂用于手性胺的合成。
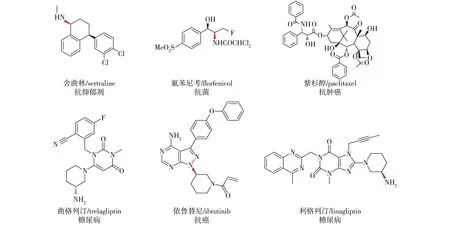
图1 含有手性胺结构的药物分子Fig.1 Pharmaceuticals contain a chiral amine moiety
生物体内错综复杂的代谢途径中含有各种各样的氧化还原酶,其中包括可以催化亚胺还原反应的酶类。如,二氢叶酸还原酶(DHFR),其在叶酸的代谢途径中可以将二氢叶酸还原成四氢叶酸(图2)[1],后者作为一碳基团的载体参与到嘧啶与嘌呤的合成,进而进一步合成核酸。这种能够催化亚胺还原的酶对于生物的正常代谢活动至关重要。然而,这些可以催化亚胺还原的氧化还原酶,虽能高效催化相应的天然底物,但由于其功能上的高度专一性,导致其所能催化的底物范围十分有限,难以催化非天然的亚胺底物。
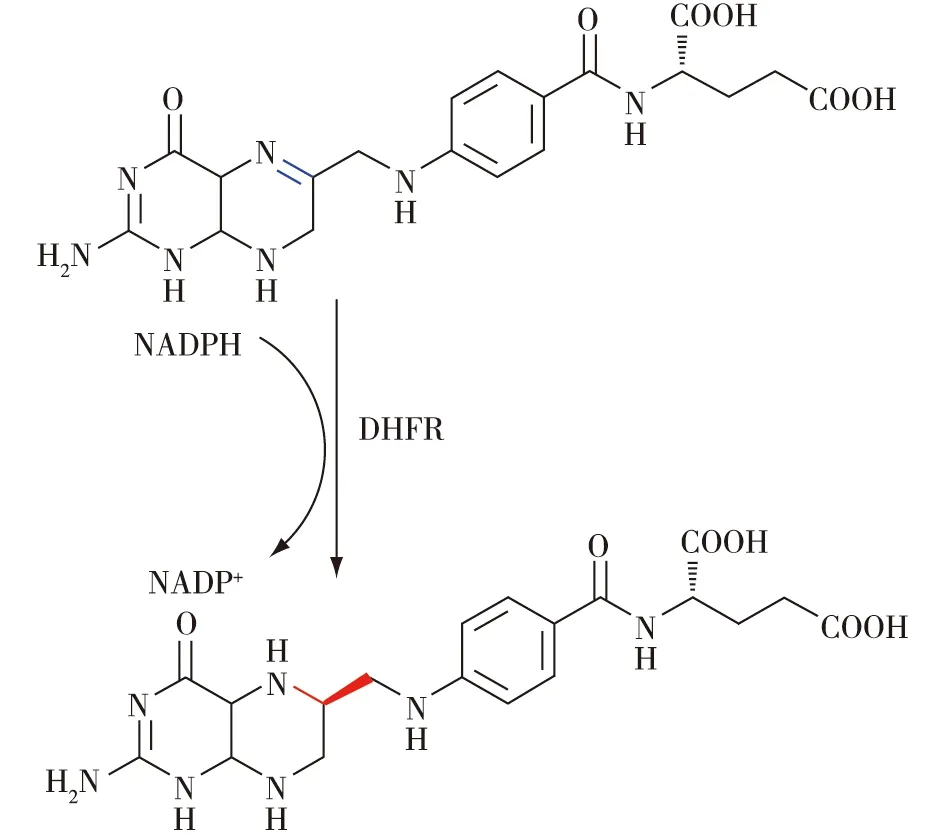
图2 二氢叶酸还原酶催化二氢叶酸还原生成四氢叶酸的反应[1]Fig.2 Reduction of dihydrofolic acid to tetrahydrogen folic acid by dihydrofolate reductase[1]
2010年,日本的Mitsukura等[2]以2-甲基-1-吡咯啉作为模式底物,筛选到2种具有亚胺还原功能的链霉菌,其中,来自Streptomycessp.GF3587 的亚胺还原酶(RIR)可以催化底物生成(R)-2-甲基吡咯烷,而来自Streptomycessp.GF3546的亚胺还原酶(SIR)可以催化底物生成(S)-2-甲基吡咯烷(图3),两种产物的光学纯度——对映体过量值(e.e.值)分别为99.2%和92.3%。Mitsukura等[3]将RIR纯化表征后发现,RIR具有严格的底物特异性,只能催化2-甲基-1-吡咯啉。而相对来说,SIR具有更宽泛的底物谱,不仅可催化2-甲基-1-吡咯啉化合物,还能催化1-甲基-3,4-二氢异喹啉和6,7-二甲氧基-1-甲基-3,4-二氢异喹啉等环状亚胺[4]。亚胺还原酶RIR和SIR的发现,不仅推动了该类酶在亚胺还原领域的迅速发展,而且还不断拓展和丰富了其催化的底物谱和反应类型。
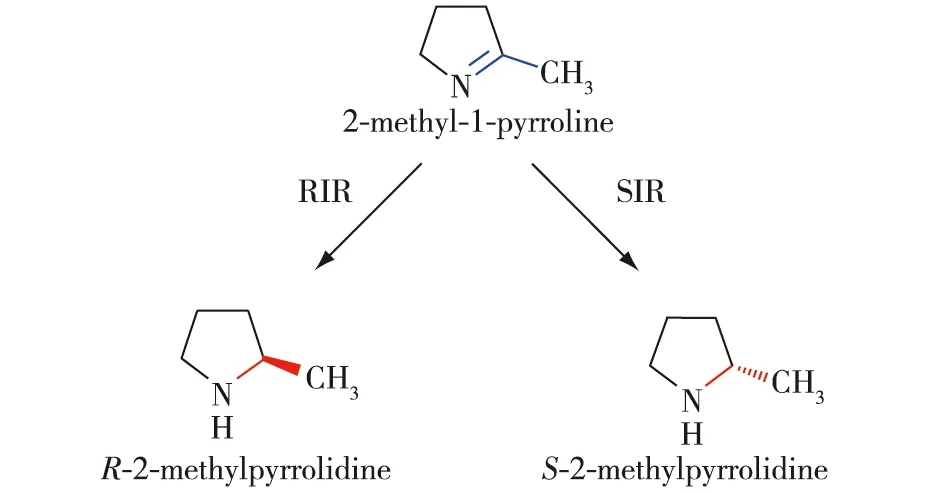
图3 亚胺还原酶SIR和RIR催化的2-甲基-1-吡咯啉的不对称还原[2]Fig.3 Asymmetric reduction of 2-methyl-1-pyrroline by imine reductases SIR and RIR[2]
1 亚胺还原酶催化的亚胺还原
2013年,Leipold等[5]发现,来自Streptomycessp.GF3546的亚胺还原酶SIR具有更广的底物谱,可以高效不对称还原五元、六元和七元环状亚胺;除此以外,SIR还可以还原亚胺离子和一些大位阻亚胺,如二氢-β-咔啉和二氢异喹啉类底物(图4)。
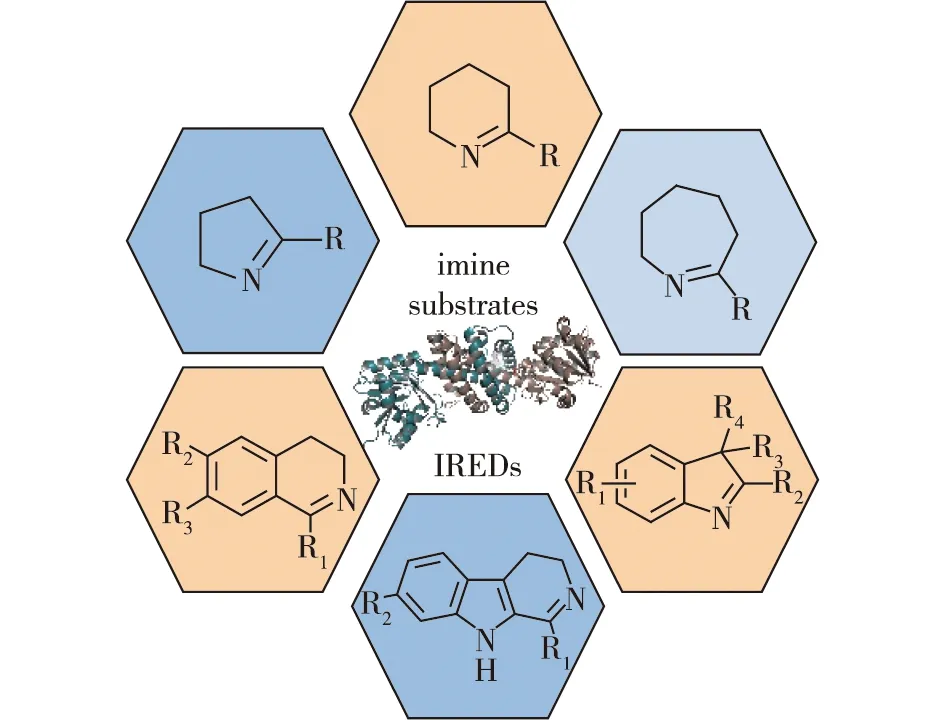
图4 亚胺还原酶催化的亚胺底物谱[5]Fig.4 The substrate scopes of imine reductases[5]
2015年,笔者所在课题组的Li等[6-7]筛选到一株乳酸菌Paenibacillus lactis来源的亚胺还原酶PlSIR(图4),可以高效不对称还原3H-吲哚及其衍生物,且e.e.值大于99%。随后,Li等[8]又筛选到对四氢异喹啉类底物具有较高催化活性的亚胺还原酶SnIR(来源于Stackebrandita nassauensis)(图4),其可催化四氢异喹啉类底物为S构型的产物,底物的转化率和产物的e.e.值均大于99%。近期,Zhang等[9]筛选获得了能够催化2-芳基吡咯啉底物的天然亚胺还原酶。针对天然亚胺还原酶催化2-芳基吡咯啉类化合物活性较低的问题,Chen等[10]通过定向进化与反应过程强化成功实现了广谱抗肿瘤药物拉罗替尼(larotrectinib)中间体的工业化生产,时空产率高达352 g/(L·d),e.e.值>99.5%,转化率>99%(图5),这证实了亚胺还原酶在亚胺还原合成中的应用潜力。

图5 工程化亚胺还原酶ScIRED催化合成拉罗替尼手性中间体[10]Fig.5 Production of the chiral larotrectinib intermediate using engineered ScIRED[10]
Wetzl等[11]以不同的单环、双环脂肪族亚胺以及具有苯基官能团的芳香族底物为模式底物,表征15个候选亚胺还原酶的催化能力,进一步拓宽亚胺还原酶环状亚胺底物谱。2016年,Aleku等[12]鉴定了一种拟无枝酸菌属Amycolatopsis orientalis来源的亚胺还原酶AoIRED (imine reductase,IRED),其可以催化合成2-取代吡咯烷、2-取代哌啶、2-取代环己亚胺和1-甲基四氢异喹啉化合物(图6)。

图6 亚胺还原酶AoIRED催化的亚胺还原[12]Fig.6 Biocatalytic reduction of imines by AoIRED[12]
2017年,Qu等[13]利用1-芳基-二氢异喹啉化合物作为筛选底物,获得了一系列对大位阻二氢异喹啉类化合物具有优良催化效率和立体选择性的亚胺还原酶(图7),其中IR45保持严格的S构型偏好性,通过对其结构的观察后发现,IR45的W191位点对空间位阻影响较大,当将191位突变为较小的丙氨酸后,使得底物抑制效应解除,催化效率提高8倍。2020年,该课题组的Yang等[14]通过理性设计成功筛选得到2个催化活性较野生型提升的亚胺还原酶突变体,对于大位阻的1-芳基-6,7-二甲氧基-二氢异喹啉可实现高立体选择性地完全转化。

图7 亚胺还原酶催化的1-芳基取代四氢异喹啉化合物合成[13]Fig.7 Synthesis of 1-aryl-substituted tetrahydro-isoquinolines by imine reductases[13]
2019年,Zumbrägel等[15]利用亚胺还原酶,在温和的条件下,实现10 g/L 2H-1,4-苯并恶嗪的不对称还原,e.e.值>99%,分离得率达71%。同年,Zumbrägel等[16]用亚胺还原酶催化获得化学方法难以制备的2-取代-3-噻唑烷类外消旋化合物,底物转化率可达96%。Yao等[17]经过对48个亚胺还原酶的筛选后发现,来自Sandarearacinus amylolyticus的亚胺还原酶IR40对大位阻底物的耐受性较好,可催化多种1-苄基六氢异喹啉底物生成相应的1-苄基八氢异喹啉衍生物(图8)。
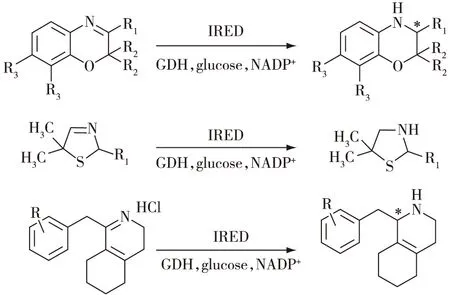
图8 亚胺还原酶催化的环状亚胺不对称还原[17]Fig.8 Asymmetric reduction of cyclicimines by imine reductases[17]
2 亚胺还原酶催化的酮还原胺化
2014年,Müller课题组的Huber等[18]首次发现来自Streptomycessp.GF3546的亚胺还原酶能够催化酮与甲胺发生还原胺化反应,尽管底物的转化率只有8.8%(图9),但该功能的发现极大地拓宽了亚胺还原酶的应用范围。随后,Wetzl等[19]通过筛选获得了能够催化酮还原胺化的2种亚胺还原酶,以此实现(1S,3R)-3-二甲基环己胺和(R)-N-甲基-2-氨基己烷的制备,具有良好得率(71%、55%)及优异的选择性(非对映体过量值(d.e.值)为98%、e.e.值为96%)。

图9 亚胺还原酶催化的酮与甲胺之间的还原胺化反应[18]Fig.9 Reductive amination of ketone and methylamine by imine reductase[18]
2017年,Aleku等[20]通过序列比对,获得了一个来自米曲霉的酶催化剂(AspRedAm),通过4组羰基底物与三组胺供体之间的还原胺化反应测定,证实了该酶的还原胺化功能及广泛的底物谱,并将其命名为还原胺化酶(图10)。随后2021年该课题组的Marshall等[21]又通过宏基因组挖掘策略鉴定了384种亚胺还原酶,并开发了一种快速简单的显色高通量筛选方法来鉴定具有酮还原胺化能力的亚胺还原酶。该筛选方法不仅可鉴定催化位阻较大酮的亚胺还原酶,还能鉴定耐热型亚胺还原酶,这项工作极大地扩展了亚胺还原酶的应用范围。

图10 还原亚胺酶AspRedAm催化的还原胺化反应[20]Fig.10 Reductive amination of ketones and amines catalyzed by AspRedAm[20]
由于亚胺还原酶在酮还原胺化研究中的重大突破,人们开始探索其在工业应用中的可行性。葛兰素史克公司的Schober等[22]利用亚胺还原酶催化的不对称还原胺化来合成LSD1抑制剂GSK2879552(图11),对天然亚胺还原酶进行3轮进化,获得了一个具有13个突变位点的最优变体,转化数(TON)比野生型提高了38 000倍;在pH 4.6的缓冲介质中反应,底物醛上载量达16.6 g/L,催化剂用量仅为1.2%(质量分数),底物转化率为91.4%,产物分离收率达到72.2%,e.e.值为99.7%;随后,在20 L规模的制备反应中,产物分离收率提升到84.4%,纯度大于99.9%,e.e.值高达99.7%,首次实现了亚胺还原酶在制药中的应用。

图11 亚胺还原酶催化合成赖氨酸特异性去甲基化酶-1抑制剂GSK2879552[22] Fig.11 Synthesis of lysine-specific demethylase-1 inhibitor GSK2879552 by imine reductase[22]
2021年,辉瑞公司的Kumar等[23]利用亚胺还原酶催化的环酮和甲胺还原胺化合成药物abrocitinib中间体(图12):通过计算和生物信息学相结合的方式对天然亚胺还原酶进行3轮酶工程改造,获得的最优突变体SpRedAm-R3-V6 (N131H/A170C/F180M/G217D),它的催化性能比天然酶的提升206倍;在百千克规模的试验中,反应48 h底物转化率高达92.5%,产物分离收率达到73%,时空产率为60 g/(L·d),纯度大于99.5%、两个非对映异构体的比(d.r.值)大于99∶1,TON高达36 538,从而实现了JAK1抑制剂abrocitinib关键中间体的商业化生产。
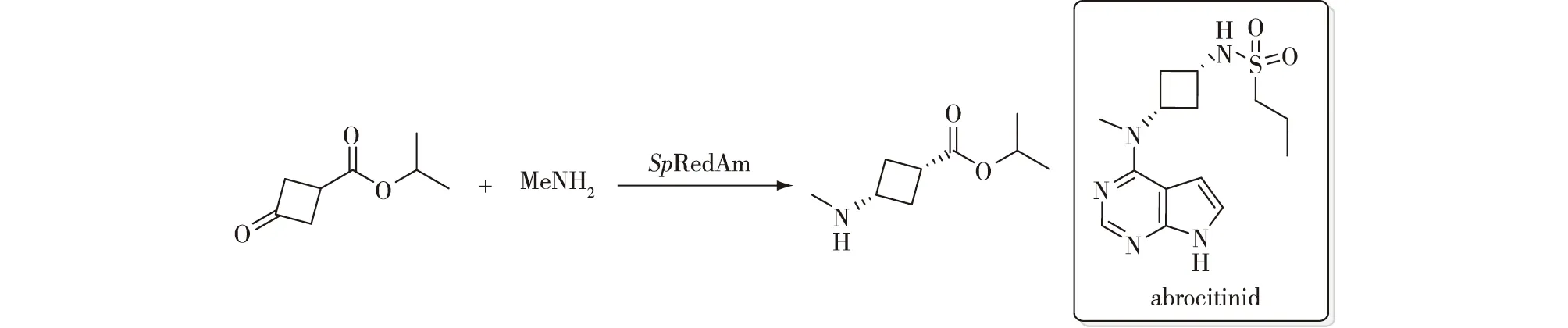
图12 亚胺还原酶突变体催化生产abrocitinib中间体[23]Fig.12 Synthesis of abrocitinib intermediate using engineered imine reductase[23]
随后,Yao等[24]利用宏基因组策略获得的亚胺还原酶催化脂肪族和芳香族α-酮酯的还原胺化反应,合成N-取代α-氨基酸酯(图13):以2-氧代-4-苯基丁酸乙酯与炔丙胺作为模式底物,对宏基因组文库鉴定出的384种亚胺还原酶进行了筛选后发现,有99种亚胺还原酶对酮酯类化合物具有催化活性,其中78种亚胺还原酶生成R构型产物,其他20种亚胺还原酶生成S构型产物;对其中催化活性较好的12种亚胺还原酶,进行了多种脂肪族和芳香族α-酮酯与不同有机胺供体之间还原胺化活性的测定;利用不同选择性的亚胺还原酶催化制备了不同构型的N-取代α-氨基酸酯,转化率为53%~99%,分离收率达27%~80%,其中R-选择性的亚胺还原酶催化产物的e.e.值为98%~99%,S-选择性亚胺还原酶催化产物的e.e.值为26%~99%。

图13 亚胺还原酶催化还原胺化不对称合成N-取代α-氨基酸酯[24]Fig.13 Asymmetric synthesis of N-substituted α-amino esters by imine reductase-catalyzed reductive amination[24]
2022年,Thorpe等[25]从鉴定的宏基因组亚胺还原酶库中,筛选出了44种具有双催化功能的亚胺还原酶(EneIRED),它们不仅能够催化羰基的还原胺化,而且能够催化C=C双键的还原(图14)。多功能酶EneIRED的发现,为多手性胺化合物的合成提供一条更简单、高效、绿色的路线。

图14 多功能亚胺还原酶催化的烯烃还原与羰基还原胺化偶联反应[25]Fig.14 Multifunctional biocatalyst EneIRED for conjugate reduction and reductive amination[25]
手性氮杂环化合物是许多手性药物的重要中间体,如治疗糖尿病药物利拉利汀(linagliptin)和阿洛列汀(alogliptin)、治疗淋巴瘤药物依鲁替尼(ibrutinib)以及治疗类风湿性关节炎药物托法替尼(tofacitinib)等[26-29]。Zhang等[30]利用亚胺还原酶催化氮杂环酮与有机胺供体合成烷基化手性氮杂环化合物(图15):以N-Boc-3-哌啶酮和苄胺为模式底物,从86 种IRED酶库中筛选出9种具有催化活性的酶催化剂;针对天然酶IR-G36催化效率极低和立体选择性不足的问题,对酶进行分子改造,获得了催化效率比母本提高了4 193倍的最优突变体M5 (kcat/Km为3.350 L/(mmol·min)),同时也显著提高了该酶的热稳定性(Tm值提高16.2 ℃);对模式底物的克级制备反应中,当底物投入量为24.9 g/L时,产物转化率达到97%、时空产率高达35.3 g/(L·d)。利用该突变体,实现了不同氮杂环酮与多种有机胺供体的直接还原胺化制备反应。

图15 亚胺还原酶催化氮杂环酮的还原胺化反应[30]Fig.15 Asymmetric synthesis of azacycloalkyl amines via imine reductase-catalyzed reductive amination[30]
Chen等[31]发现,青霉菌Penicillium camemberti来源的亚胺还原酶PcIRED能实现羰基底物与大位阻胺之间的还原胺化反应,但是天然酶PcIRED催化模式底物3-(3-三氟甲基苯基)丙醛与(R)-1-(1-萘基)乙胺还原胺化合成药物西那卡塞(cinacalcet)的转化率仅为0.63%,随后他们对该酶进行了分子改造,通过3轮突变,显著提高了该酶的催化效率,最优突变体PcIRED-M3的比酶活为8.14 U/mg,比野生型提高了488倍;同时该酶的热稳定性也显著提升,其Tm值为51.5 ℃,比野生型提高了10 ℃;利用最优突变体PcIRED-M3,实现了西那卡塞的克级规模制备,底物转化率为93%,e.e.值为99%,产物分离得率高达 85% (图16)。最后,还利用突变体与野生酶分别研究不同羰基底物与大位阻胺供体之间的还原胺化反应后发现,突变体能够实现多种大位阻胺类药物分子或药物前体的酶促合成。如,钙敏受体激动剂替卡塞(tecalcet)[32]、灭虫剂苄酚宁(bephenium)药物前体[33]、α-肾上腺素受体拮抗剂酚苄明(phenoxybenzamine)药物前体[34]、5-HT1A受体拮抗剂阿尔维林(alverine) 药物前体[35]以及抗真菌萘替芬(naftifine) 药物前体[36]。
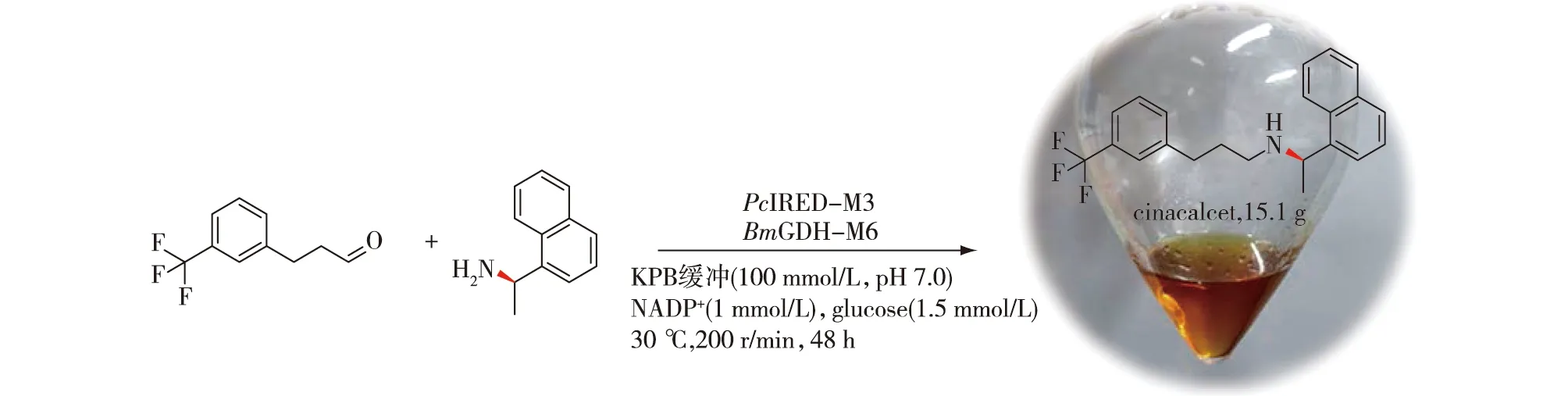
图16 PcIRED-M3催化的西那卡塞合成反应[31]Fig.16 Preparative synthesis of cinacalcet catalyzed by PcIRED-M3[31]
3 涉及亚胺还原酶催化的级联反应
多酶级联反应在生物和化学合成方面受到越来越多的关注,这是因为多酶级联反应无须基团的保护和脱保护,不需要苛刻的反应条件,可以产生化学方法难以获得的高官能团和高光学纯度的手性分子。在以前研究工作[37-38]中,利用IRED对亚胺中间体的选择性还原取代了胺氧化酶和非选择性化学还原剂级联,通过级联葡萄糖脱氢酶,额外添加葡萄糖就可以使得NADPH辅酶再生。基于IREDs的多酶级联体系开发为取代的N-杂环吡咯烷和哌啶提供了有效的生物合成途径。这些手性取代的五元和六元N-杂环化合物存在于生物活性产物和药物分子中。
Heath等[39]构建了6-羟基-D-尼古丁氧化酶(6-HDNO)与IRED级联的多酶催化体系,实现了N-杂环化合物的去外消旋化(图17):先利用6-HDNO突变体将外消旋吡咯烷和哌啶的一个对映体去消旋化,生成相应的亚胺中间体,随后利用S选择性的IRED进行不对称还原,生成单一构型的手性胺化合物,转化率可以达到71%~99%。

图17 亚胺还原酶和胺氧化酶级联催化N-杂环的去消旋化[39]Fig.17 Deracemization of nitrogen heterocycles by imine reductase and amine oxidase[39]
France等[40]和Hepworth等[41]通过羧酸还原酶(CAR)、ω-转氨酶(ω-TA)和IRED的级联(图18),实现了从酮酸或酮醛到手性单取代和双取代哌啶和吡咯烷的生物催化合成:首先来自海洋分枝杆菌的CAR,将酮酸还原为酮醛;然后,再利用转氨酶ATA-113催化醛胺化生成氨基酮,并自发环化形成环状亚胺化合物;最后,形成的亚胺分别被立体选择性互补的IRED进行不对称还原,获得两种对映异构体的胺产物。该系统还构建了辅因子自循环系统,实现了辅因子的自给自足,通过体内外级联反应,合成了系列高立体选择性(e.e.值和d.e.值都大于98%)的单取代/双取代哌啶和吡咯烷。
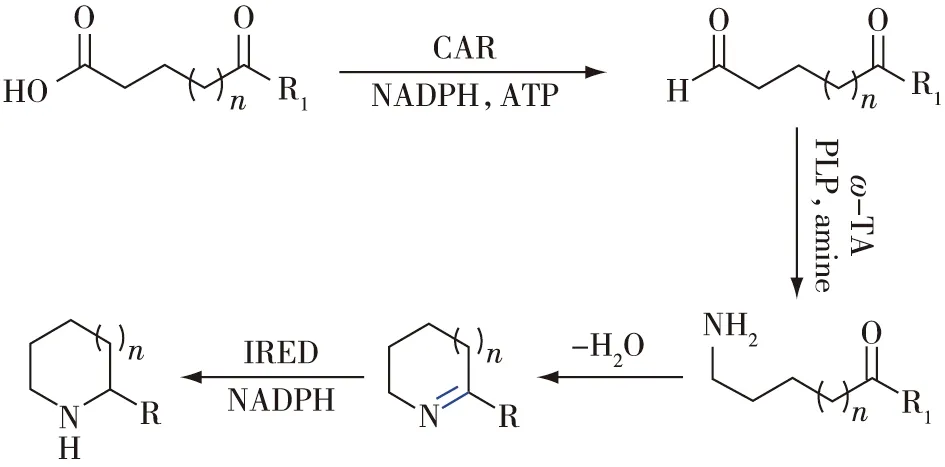
图18 羧酸还原酶、ω-转氨酶和亚胺还原酶一锅法级联合成哌啶和吡咯烷[40-41]Fig.18 One-pot cascade synthesis of piperidines and pyrrolidines using carboxylic acid reductase,ω-transaminase and imine reductase[40-41]
Slabu等[42]构建了大肠杆菌K12来源的腐胺转氨酶pATAs与IRED级联的反应,实现了从链状二胺到氮杂环化合物的合成(图19):首先利用腐胺转氨酶与氨基酸氧化酶级联将腐胺和尸胺的其中一个胺基转化为醛基,醛基胺中间体经自发环化形成1-吡咯啉或1-哌啶;然后,经IRED催化的立体选择性还原生成相应的吡咯烷和哌啶产物,该途径可以实现99%的底物转化率。
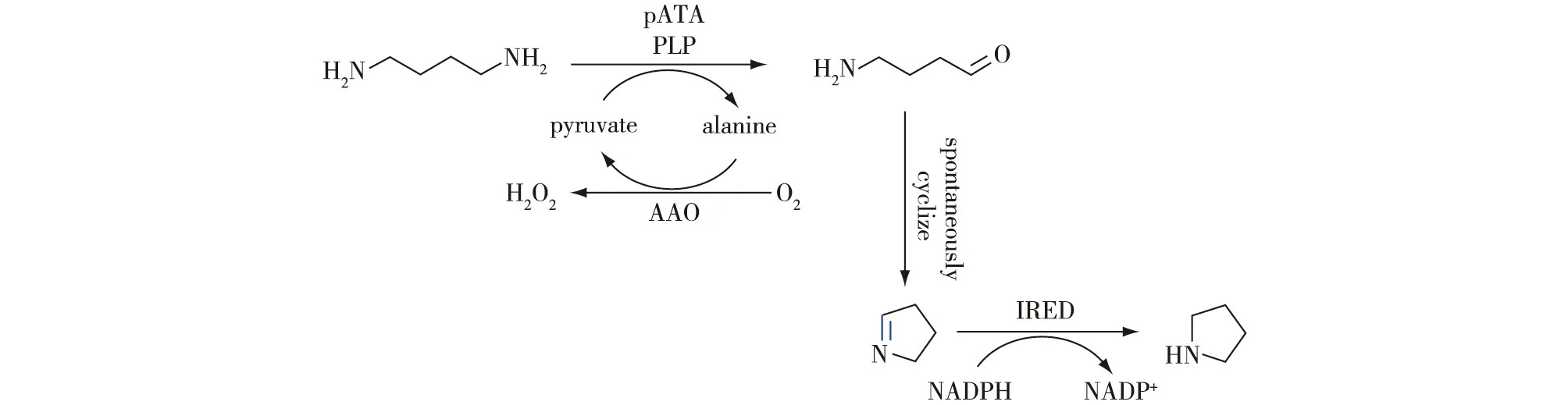
图19 腐胺转氨酶和亚胺还原酶级联催化二胺生成饱和N-杂环化合物[42]Fig.19 Synthesis of saturated nitrogen heterocycles from diamines using putrescine transaminases and imine reductase[42]
在此基础上,Borlinghaus等[43]对此级联反应进行了优化,利用工程化的腐胺氧化酶(PuO)和IRED进行级联,实现了从二胺到N-杂环吡咯烷和哌啶的合成(图20):首先,对来自红串红球菌的腐胺氧化酶进行工程改造,并利用工程化的腐胺氧化酶对取代的二胺进行选择性氧化;然后,通过IREDs催化的亚胺中间体不对称还原,形成相应的N-杂环化合物。通过该双酶级联反应,可以实现从1,5-二氨基-2-甲基戊烷到3-甲基哌啶的高效合成。

图20 腐胺氧化酶和亚胺还原酶全细胞级联催化制备3-甲基哌啶[43]Fig.20 Putrescine oxidase and imine reductase cascade biotransformation to access 3-methylpiperidine in whole cells[43]
Thorpe等[44]构建了烯烃还原酶(ERED)与IRED级联的多酶催化体系,首先通过催化ERED烯胺(α,β-不饱和亚胺)中 C=C键还原,然后再通过IRED介导的C=N还原,制备非对映异构富集的2-取代饱和N-杂环(图21)。利用该途径,对手性胺(S)-2-(1 -甲基乙基)哌啶和(RS)-2-仲丁基哌啶进行了制备合成,得到具有优异对映选择性(e.e.值>99%)的盐酸盐产物。
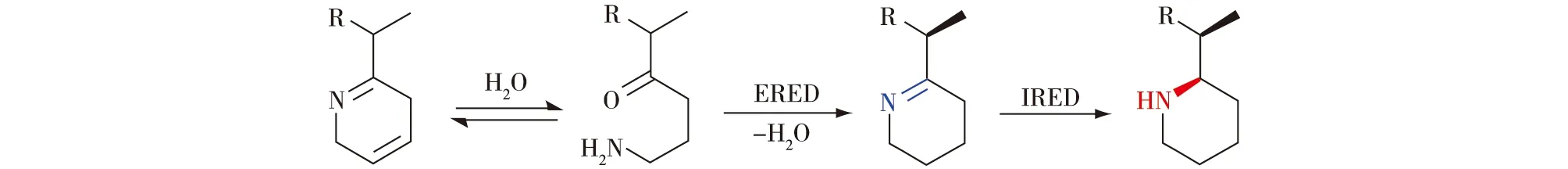
图21 烯烃还原酶和亚胺还原酶一锅级联催化还原环亚胺制备饱和N-杂环化合物[44]Fig.21 One-pot biocatalytic cascade reduction of cyclic enimines to N-heterocycles by olefin reductase and imine reductase[44]
随后,Harawa等[45]构建了化学催化与生物催化级联的化学-酶法合成体系,使用非对映选择性的一锅胺氧化酶/烯亚胺还原酶级联反应,将N-取代的四氢吡啶(THPs)转化为手性的3-和3,4-取代的哌啶(图22):使用NaBH4作为氢化物源,催化活化的吡啶生成相应的THPS,通过级联胺氧化酶(AmOx)原位催化氧化THP生成相应的二氢吡啶(DHPs),产生与C=N键偶联的活化C=C键,然后被双功能酶EneIREDs还原,生成3-和3,4-取代哌啶。

图22 胺氧化酶和亚胺还原酶级联催化合成手性哌啶[45]Fig.22 Synthesis of chiral piperidine by amine oxidase and imine reductase[45]
Mattey等[46]采用多点注射反应器(MPIR)开发了一种连续流动系统(图23),并使用工程化胆碱氧化酶 AcCO6生成醛来测试MPIR的可行性;再将固定化转氨酶BmTA后装入填充床后连接到MPIR的输出端产生MPIR填充床系统(MPBS),实现伯胺2的高效合成。同样,利用AdRedAm与BsGDH级联反应实现了仲胺3的高效合成。
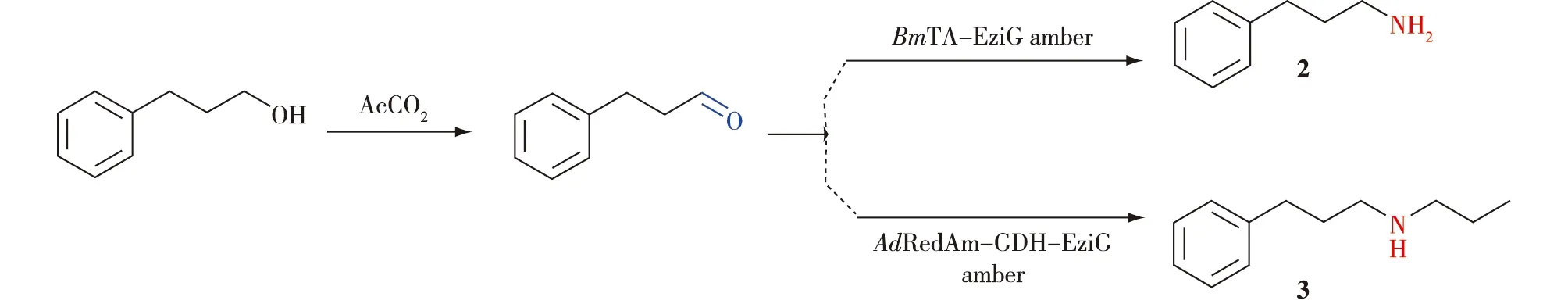
图23 胆碱氧化酶和亚胺还原酶级联催化合成仲胺的连续流系统[46]Fig.23 Continuous flow systems to access secondary amines by chemoenzymatic cascade[46]
近期,瞿旭东课题组的Zhu等[47]对亚胺还原酶进行了分子改造,实现了一系列邻位、间位、对位及多取代(S)-1-苯基-THβCs的高效合成(图24):通过级联一个N-甲基转移酶CNMT,将14种不同取代基的1-苯基- THβCs完全转化为相应的N-甲基化产物;用S-腺苷-L-腺苷同型半胱氨酸(SAH)替代S-腺苷甲硫氨酸(SAM)作为甲基基团的供体参与转甲基反应,通过腺苷蛋氨酸循环再生SAM,实现一锅法合成(S)-N-甲基苯基-THβCs。

图24 三酶级联一锅法生成N-甲基化1-苯基-THβCs[47]Fig.24 Synthesis of 1-aryl-tetrahydro-β-carbolines by imine reductase and methyltransferases[47]
4 亚胺还原酶的结构及反应机制
蛋白的三维结构是理解酶催化机制的基础,同时也是对酶进行理性改造的起点。Grogan 课题组的Rodríguez-Mata等[48]于2013年解析了第一个亚胺还原酶的蛋白结构 (PDB:3ZHB)以来,截至目前,已有数十个亚胺还原酶的结构被陆续解析。亚胺还原酶的三维结构呈现同源二聚体模式,与β-羟基酸脱氢酶三维结构相似,其中,蛋白的N端为Rossmann折叠,C端为α螺旋束,N端和C端之间由一条长的α螺旋连接(图25(a)),两条单体之间相互交叉形成一个结构空腔(图25(b))。与其他NAD(P)H依赖的氧化还原酶一致,由位于N端的Rossmann折叠为辅酶NADPH提供结合口袋;与β-羟基酸脱氢酶所不同的是,亚胺还原酶的底物结合口袋位于由A链N端的Rossmann折叠与B链C端的α螺旋束通过交叉形成的空腔中[48]。

图25 亚胺还原酶晶体结构示意[48]Fig.25 The cartoon style of protein structure of imine reductase[48]

在反应过程中,Asp187对亚胺离子中间体起稳定以及质子化的作用,随后该亚胺离子中间体被来自辅酶烟酰胺环上C4位的氢原子还原为胺产物图26 推测亚胺还原酶催化机制示意[48]Fig.26 Speculative reaction mechanism of imine reduction(CR) [48]
探究酶催化机制是生物催化研究不可或缺的部分,清晰的催化机制不仅可以揭示酶功能背后神秘的构效关系,同时也可以为酶工程提供重要的参考信息。Rodríguez-Mata等[48]通过与催化机制已被阐述清楚的酮还原酶 (PDB:2CVZ)进行结构比对,进而推测亚胺还原酶(PDB:3ZHB)中的Asp187在亚胺还原反应中扮演着催化残基的角色(图 26)。基于此,他们将Asp187突变成Ala187或Asn187后发现,2个突变体均不再表现出亚胺还原活性。该研究似乎验证了Asp作为催化残基的推测,但是随后越来越多的研究发现,将其他亚胺还原酶对应的该位点残基突变成Ala、Gly及Asn等残基时,酶依然具有还原亚胺的能力。不仅如此,在该残基位点上是Ala或者Asn的新酶也具有亚胺还原活性[12]。进一步通过分析酶与NADPH复合物发现,假定的催化残基Asp187与NADPH的C4原子之间的距离超过了0.4 nm,超出了形成氢键的距离[48-50]。此外,分析酶-产物-NADPH的三元复合物结构发现,假定的催化位点与氨基-NADPH形成的中间体的距离更远,如Asn171(对应PDB:3ZHB中的Asp187)与芳香族产物之间的距离约为0.5 nm[12]。上述这些结构上的现象均有悖于传统的氧化还原酶催化机制。因此,将Asp187视作整个亚胺还原酶家族唯一的催化残基这一机制是不完善的。基于这一背景,随后提出了其他的催化机制假说,如基于二氢叶酸脱氢酶所提出的氢键交互网络催化假说[49,51]。虽然已有多个亚胺还原酶结构已被解析,但是距离完全将其催化机制阐述清楚还任重而道远。综上可知:①亚胺还原酶可能具有更为复杂的催化机制;②亚胺还原酶具有多样性的催化机制。
5 人工亚胺还原酶
截至目前,尽管已有大量自然亚胺还原酶被挖掘表征,不过一些天然酶“与生俱来”的缺陷 (如,较差的长期稳定性、有限的底物谱、昂贵的辅因子循环系统) 使得人工亚胺还原酶仍然具有潜在的研究前景[52]。由于金属催化具有十分广谱的催化范围,因此在有机合成中被广泛使用。基于此,近十几年来已有不少研究者尝试将多种金属构建到蛋白骨架中,使其以金属酶的角色执行催化亚胺还原的功能。相较于单纯的金属催化,构建金属亚胺还原酶具有4个优点[51]:①金属酶催化可在温和的反应条件下进行;②蛋白骨架可保护金属的催化能力不受外界环境的破坏;③金属酶通常具有较高的催化效率;④金属酶具有更高的立体选择性。
多种蛋白质骨架已被用于构建金属亚胺还原酶,最为常见的有链霉亲和素、核糖核酸酶S、碳酸酐酶以及周质结合蛋白[53-56]。Dürrenberger等[53]将芳烃钌通过双齿配体将其整合到链霉亲和素骨架中可以实现其在亚胺还原反应中立体选择性的从无到有。碳酸酐酶由于其在大肠杆菌中具有高表达的优点,因此也常被当作构建人工金属亚胺还原酶的重要蛋白载体。如Raines等[56]将具有催化不对称加氢反应IrCp*有机金属催化剂整合进碳酸酐酶骨架中,成功构建出一种金属亚胺还原酶。由此可见,作为天然亚胺还原酶的有利补充,人工金属酶在亚胺还原研究领域也具有一定的研究潜力。
6 总结与展望
近年来,亚胺还原酶的研究取得了巨大的进展,大量的天然亚胺还原酶已经被鉴定,其催化功能不断得到拓展,不仅可以用于催化亚胺的还原,还可以催化酮的还原胺化。但是相对转氨酶、胺氧化酶等合成手性胺的酶系,对于该类酶的研究仍处于初始阶段,目前高活性的天然亚胺还原酶还鲜有报道,亚胺还原酶的催化机制和天然功能还不清晰。未来在新酶的开发、酶催化混杂性、酶的分子改造、酶的催化机制解析、电酶催化等方面还值得深入研究和探索。
此外,该类酶的大规模工业应用存在着极大的挑战,需要解决活性低、立体选择性不足、底物抑制等问题,需要借助定向进化技术和工程强化手段来解决这些限制。在实际应用中除了活性、稳定性和选择性等酶学性质表征外,对生产性能表征也至关重要。底物加载量、催化剂加载量、产物得率和产物光学纯度等生产性能可以更好反映酶催化剂的应用潜力。
