羟基酪醇柔性纳米脂质体的制备及其贮藏稳定性研究
2023-10-09李文君王成章雷建都陈虹霞
李文君, 王成章, 雷建都, 陈虹霞
(1.中国林业科学研究院 林产化学工业研究所;江苏省生物质能源与材料重点实验室;国家林业和草原局林产化学工程重点实验室;林木生物质低碳高效利用国家工程研究中心;江苏省林业资源高效加工利用协同创新中心,江苏 南京 210042; 2.北京林业大学 材料科学与技术学院,北京 100083; 3.南京林业大学江苏省林业资源高效加工利用协同创新中心,江苏 南京 210037)
羟基酪醇(HT),是油橄榄中的主要酚类化合物,可以从橄榄叶和橄榄油中提取,也可以由橄榄苦苷水解生成[1]。HT具有抗氧化、抗炎、抗动脉粥样硬化、抗血栓形成、改善内皮功能障碍、脂质和止血功能等[2]。此外,HT还具有抗肿瘤活性,可作为神经保护剂[3]、心脏保护剂[4]和化学预防剂[5],用于潜在安全的药物、天然食品添加剂和化妆品成分中。但是,HT因含有3个羟基,对空气和光非常敏感,具有很强的不稳定性和亲水性,从而影响了其生物活性,使其易于快速释放并在体内降解,导致了其较低的生物利用度[6]。而脂质体作为药物递送载体,具有靶向性、缓释性和保护性等许多提高药物有效性的优良特性[7-11],如抗肌氨酸的抗体叶酸靶向脂质体,表现出竞争性抑制作用[12];与游离药物相比,药物从脂质体中释放慢,作用时间延长[13],且在心脏和肾脏中的蓄积显著降低[14],并由于脂质体的保护,其稳定性增加[15]。氢化卵磷脂和胆固醇具有良好的生物相容性和生物降解性,用作脂质体壁材料,能够通过提高药物溶解度、生物利用度和体内外稳定性以及防止干扰来增强生物活性。柔性脂质体由磷脂和能促使磷脂的双分子层动摇和变形的边缘活化剂(如胆酸钠)组成[16],边缘活化剂的存在扰乱了脂质体的磷脂双分子层结构,增加了膜的流动性和柔性,使得柔性脂质体(甚至是较大尺寸)在角质层细胞间挤压以及体内经皮渗透压梯度作用下发生变形而透过完整的皮肤[17-18]。近年来,已有报道柔性脂质体作为载体促进胰岛素、疫苗、非类固醇类(布洛芬)等药物的输运[19],其不仅能促进药物透过皮肤,也能运载这些药物直接到达皮肤下的深部软组织[20]。因此,本研究通过薄膜分散法对HT脂质体包封,研究影响脂质体形成和乳化的参数,并通过响应面优化处理,确定较佳工艺条件,同时考察其在不同温度下贮藏不同时间的稳定性,为HT柔性纳米脂质体的进一步应用奠定基础。
1 实 验
1.1 原料、试剂与仪器
羟基酪醇(HT)标准品(纯度≥98%)、氢化卵磷脂、卵磷脂、胆固醇、胆酸钠、叶酸(FA),N,N′-羰基二咪唑(CDI),上海阿拉丁生化科技股份有限公司;聚醚F127,上海麦克林生化科技有限公司;叶酸-聚醚(FA-F127),实验室自制[21];氯仿、甲酸、乙醇,均为分析纯;甲醇、乙腈、水,均为HPLC级。
LC-20T高效液相色谱仪,日本岛津公司;Nano ZS激光粒度分析仪,英国马尔文公司;JY92-IIN超声波细胞破碎仪,上海精其仪器有限公司;TGL-16M台式高速冷冻离心机,上海卢湘仪离心机仪器有限公司;RE-3000A旋蒸蒸发器,上海亚荣生化仪器厂;NICOLET IS50傅里叶变换红外光谱仪,美国赛默飞公司;TG209F1热重分析仪,德国Netzsch公司;DSC8000差示扫描量热仪,美国PerkinElmer公司;H-7650透射电子显微镜,日本日立公司。
1.2 羟基酪醇分析方法建立
1.2.1HPLC色谱条件 色谱柱为SUPELCOSILTMLC-18(25 cm× 21.2 mm, 5 μm),流动相A为1%甲酸溶液,流动相B为5%甲醇和1%甲酸的乙腈溶液,流速0.8 mL/min,进样量5 μL,梯度洗脱,以80%A开始洗脱,7 min内流动相B调整为30%并保持18 min,之后10 min内增加流动相B到95%,并保持5 min,最后调节流动相B至20%并保持5 min,总运行时间45 min。
1.2.2HPLC标准曲线的绘制 精密称取HT标准品,分别配制成质量浓度为2、 1、 0.5、 0.1、 0.05、 0.02、 0.01和0.005 g/L的标准溶液,甲醇溶解定容、摇匀,经0.45 μm的滤膜过滤后,依次进样5 μL检测(进样3次),以峰面积积分值的平均值为纵坐标(y),标准溶液质量浓度为横坐标(x),绘制标准曲线并拟合回归方程。得到HT标准曲线回归方程为:y=5 366 853x-41 226,R2=0.993 0。
1.3 羟基酪醇柔性纳米脂质体制备及其制备工艺优化
1.3.1单因素试验 采用薄膜分散法制备羟基酪醇柔性纳米脂质体,首先准确称取一定比例(质量比)的氢化卵磷脂与胆固醇于100 mL圆底烧瓶中混合,加入适量氯仿充分溶解后,加入一定质量的HT,利用旋转蒸发仪在42 ℃下浓缩去除溶剂氯仿,待圆底烧瓶内部形成白色均匀的涂层即可;然后量取3 mL胆酸钠水溶液及适量FA-F127在60 ℃下水化1 h(20 r/min);再将所得乳液通过超声波破碎处理,形成均匀的脂质体纳米乳液。
实验中分别考察HT用量(0.6、 0.9、 1.2、 1.5和3.0 mg)、氢化卵磷脂与胆固醇质量比(1∶1、 2∶1、 3∶1、 4∶1和5∶1)、FA-F127用量(0.1、 0.5、 1、 1.5和2 mg)、胆酸钠用量(5、 10、 15、 20和25 mg)、超声波功率(5%、 10%、 20%、 30%和40%,总功率为650 W)、超声波作用时间(5、 10、 15、 20和25 min,均为超声波作用2 s,中间间隔1 s)等单因素对HT柔性纳米脂质体包封率的影响。
1.3.2Plackett-Burman试验 根据单因素试验的结果,利用Design-Expert 13软件中Plackett-Burman试验设计方法,考察上述6个因素(n=12)对HT柔性纳米脂质体包封率的影响,并筛选出具有显著影响的因素。
1.3.3响应面优化试验 在Plackett-Burman试验的基础上,依据Design-Expert 13软件中Box-Behnken中心组合设计方法,以HT纳米脂质体的包封率为响应值,选择合适的因素与水平设计响应面优化试验,确定较佳制备工艺。
1.4 羟基酪醇柔性纳米脂质体结构表征
1.4.1红外光谱分析 利用傅里叶变换红外光谱(FT-IR)仪分析,波数范围为400~4 000 cm-1,每个样品循环扫描16次,分辨率为4 cm-1。
1.4.2热重分析 热重分析在氮气保护下,从40 ℃起以10 K/min的升温速率加热到800 ℃。
1.4.3差示扫描量热法 利用差示扫描量热仪(DSC)分析,在-70 ℃下保持0.5 min,然后以20 ℃/min的升温速率升温至70 ℃,保持0.5 min,以20 ℃/min的降温速率降温至-70 ℃,保持3 min后,以20 ℃/min的升温速率再次升温至70 ℃。
1.4.4透射电镜TEM分析 透射电子显微镜(TEM)用于测定HT柔性纳米脂质体的微观结构。首先取一滴样品悬浮液滴在干燥的载玻片上,再把带有支持膜的铜网放在悬浮液的液珠上漂浮以蘸取样品,然后用滤纸吸干铜网上多余悬液,再将铜网在磷钨酸染液滴珠上漂浮,时间约1.5 min,最后再用滤纸将染液吸干即可在80 kV的加速电压下观察脂质体样品的微观形态。
1.5 羟基酪醇柔性纳米脂质体稳定性研究
1.5.1包封率测定 脂质体制剂的包封率是一个重要的定性评估特征,由添加的药物总量与脂质体中包埋的药物量计算得出。取0.5 mL的脂质体纳米乳液加入1 mL 10% Triton X-100甲醇溶液(破乳剂),超声波作用2 min(破乳,释放脂质体包埋的药物),然后以12 000 r/min离心10 min。移取上清液用甲醇定容至5 mL,利用1.2节方法测定溶液中HT含量。包封率根据式(1)计算。
(1)
式中:YE—HT纳米脂质体乳液的包封率,%;co—溶液中HT的总质量浓度,g/L;c—游离HT的质量浓度, g/L;Vo—溶液中HT的总体积,mL;V—游离HT体积,mL。
1.5.2粒径、多分散系数(PDI)及Zeta电位的测定 按优化工艺条件制备HT纳米脂质体乳液,稀释一定倍数后,在25 ℃、散射角90°条件下,采用激光粒度分析仪测定HT柔性纳米脂质体的粒径、PDI及Zeta电位值。
1.5.3贮藏稳定性 按优化工艺条件制备HT纳米脂质体乳液,分别在4、 25和60 ℃下贮藏0、 7、 14、 21和28 d后按照1.5.1和1.5.2节方法测其包封率、粒径、PDI及Zeta电位。
2 结果与讨论
2.1 HT柔性纳米脂质体制备工艺优化
2.1.1单因素试验 选择HT用量、氢化卵磷脂与胆固醇质量比、FA-F127用量、胆酸钠用量、超声波功率、超声波作用时间为考察因素,探讨各因素对HT柔性纳米脂质体包封率的影响,结果见图1。

a.HT用量HT dosage;b.m(氢化卵磷脂)/m(胆固醇) m(hydrogenated lecithin)/m(cholesterol);c.FA-F127用量dosage of FA-F127;d.胆酸钠用量dosage sodium cholate;e.超声波功率ultrasonic power;f.超声波作用时间ultrasonic time
由图1可知,HT用量过高过低都会引起HT柔性纳米脂质体包封率的下降,当HT用量为1.5 mg,即HT在脂质体溶液中质量浓度为0.5 g/L时包封率最佳。同样,其他5个因素对HT柔性纳米脂质体包封率的影响也出现了先增加后下降的趋势,分别在m(氢化卵磷脂)∶m(胆固醇)=3∶1、FA-F127用量1 mg、胆酸钠用量20 mg、超声波功率30%、超声波作用时间10 min达到最大包封率。由于Plackett-Burman试验设计需要尽量涵盖每个因素允许取值的最大空间,但又不需过大,因此,试验时可选取的HT用量范围为1.2~3 mg,m(氢化卵磷脂)∶m(胆固醇)、FA-F127质量、胆酸钠质量、超声波功率、超声波作用时间可选范围分别为2∶1~4∶1、 0.5~1.5 mg、 15~25 mg、 20%~40%、 5~15 min。
2.1.2Plackett-Burman试验设计关键因素的筛选 利用Plackett-Burman试验设计方法,考察6个因素对HT柔性纳米脂质体包封率的影响,并筛选出对其影响较为显著的因素,每个因素取最低和最高两个水平,结果如表1所示。

表1 Plackett-Burman试验设计及结果
根据表1试验设计结果,进行多元回归方程拟合和方差分析,得一次回归方程:Y=42.43+0.467 5X1+1.14X2+0.454 2X3+1.29X4+0.654 2X5+1.67X6,同时对该方程进行方差分析和显著性检验,结果如表2所示。
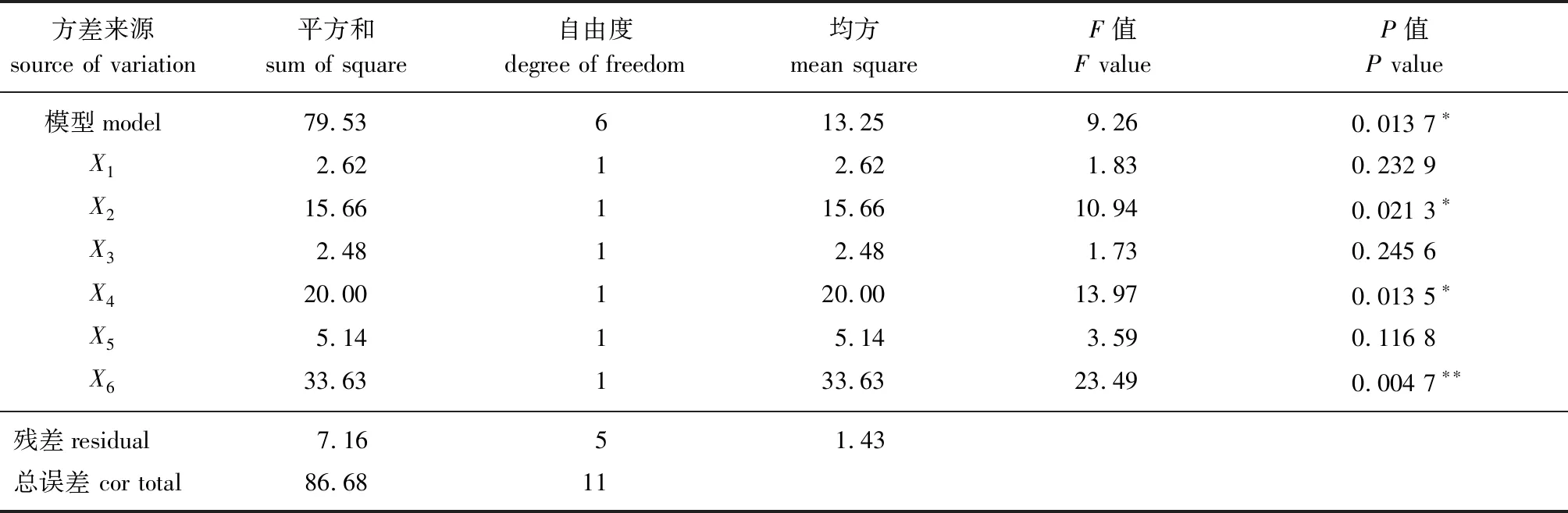
表2 Plackett-Burman试验方差分析
2.1.3Box-Behnken响应面优化 综合单因素试验和Plackett-Burman试验结果,选取超声波作用时间(X6)、胆酸钠用量(X4)和m(氢化卵磷脂)∶m(胆固醇)(X2)这3个因素为响应面模型的自变量,进行三因素三水平的Box-Behnken试验设计,以HT柔性纳米脂质体的包封率为响应值进行响应面分析。各因素水平和结果如表3所示。
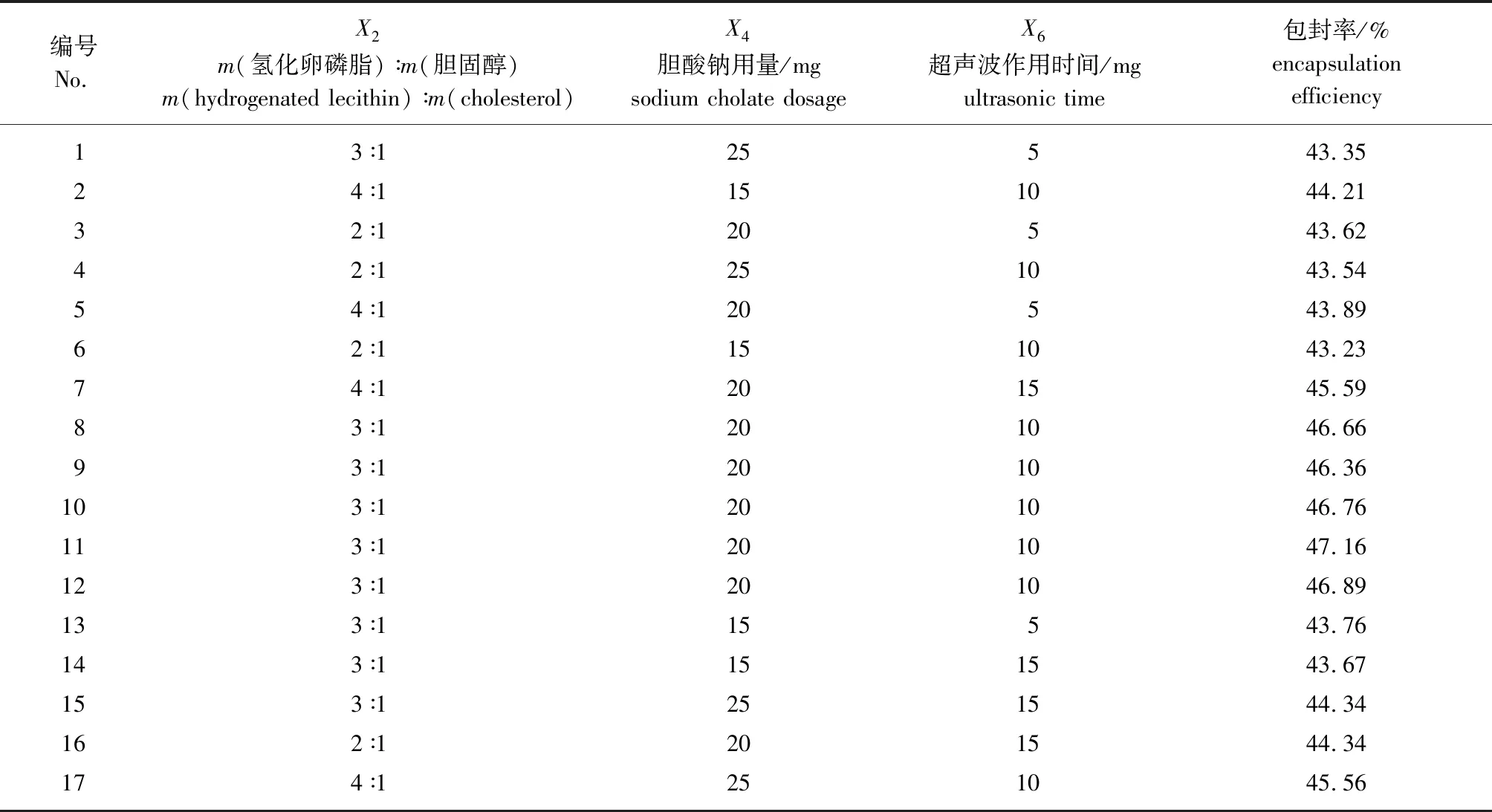
表3 Box-Behnken设计与结果

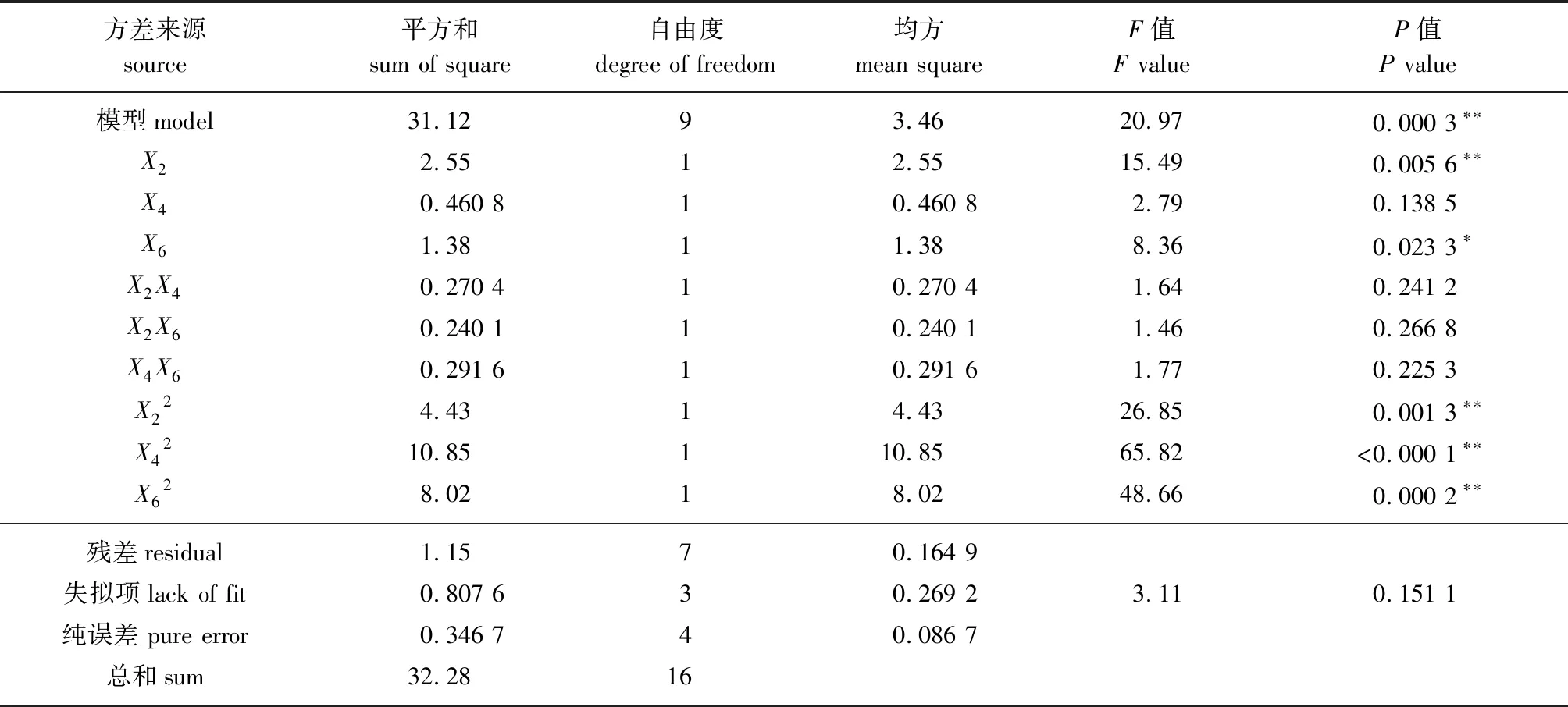
表4 回归方程方差分析
根据表4数据可知,一次项X2,二次项X22、X42、X62对HT柔性纳米脂质体包封率的影响极显著(P<0.01),X6的影响显著(P<0.05),根据F值大小可知影响包封率的因素排序为:m(氢化卵磷脂)∶m(胆固醇)(X2)>超声波作用时间(X6)>胆酸钠用量(X4)。根据模型回归方程可知,HT柔性纳米脂质体制备工艺的优化条件为m(氢化卵磷脂)∶m(胆固醇)为3.313 21∶1,胆酸钠用量为20.578 2 mg,超声波作用时间为10.944 7 min,此时HT柔性纳米脂质体包封率为46.907 6%。
为了验证上述响应面结果的可靠性,进行3次平行验证试验,考虑到实际操作,对各优化条件进行简化处理,取m(氢化卵磷脂)∶m(胆固醇)为3.3∶1,胆酸钠用量为20.6 mg,超声波作用时间为11 min,仍然选择超声波功率、HT用量和FA-F127用量分别为10%、 1.5 mg和1 mg,制备的HT柔性纳米脂质体平均包封率为46.78%,与理论值的相对误差为0.27%,说明该模型是可靠的。
2.2 HT柔性纳米脂质体结构表征
2.2.1红外光谱分析 红外光谱可通过官能团的主要波数用于确定可能的活性物质。烃链的最具特征的官能团酯基团特征峰位于1 740~1 700和1 250~1 050 cm-1之间,磷脂基团特征峰处于1 250~1 040 cm-1之间[22]。986 cm-1应该是氢化卵磷脂胆碱头的(CH3)3-N+伸缩的特征。然而,在图2(a)中,HT柔性纳米脂质体与空白脂质体(不含HT),在3 300和3 280 cm-1,986和1 008 cm-1,以及HT柔性纳米脂质体与HT在1 420和1 440 cm-1,1 000和1 100 cm-1等处的不同吸收而显示出一些变化,但是可能由于HT与脂质基质产生相容,导致载药纳米脂质体光谱中观察到载体的所有特征峰并且现有峰没有主要移动或产生出现了新的高峰[23]。

a.FT-IR;b.TG;c.DTG;d.DSC
2.2.2热重分析 样品的热重分析如图2(b)和图2(c)所示。空白脂质体和HT柔性纳米脂质体均在第一阶段表现出轻微的质量损失,这一过程主要损失的是样品中吸附的水分或者未完全除去的溶剂;在216.3 ℃后发生第二阶段主要的质量损失过程,这一过程发生了一系列复杂的C—C、C—O、酚羟基和苯环断裂等反应,由DTG曲线可知,空白脂质体和HT柔性纳米脂质体的失重率在226 ℃下分别达到-17.33 %/min和-15.028%/min,且2种脂质体最终质量损失分别为80.492%和77.78%,可能是负载了HT的脂质体增强了其热力学稳定性,这一点也可以从DTG曲线中空白脂质体有更高的失重率得到佐证。
2.2.3差示扫描量热法 HT柔性纳米脂质体的DSC分析如图2(d)所示。
由图可知,第一次升温曲线在48.52 ℃处出现一个明显的吸热峰峰值,只有一个峰值可能是因为化合物的均质化及其结晶状态转化为分散无定形状态[23],且其玻璃化转变温度Tg为-35.11 ℃。第二次升温曲线的Tg在-36.54 ℃处,2次升温曲线的Tg值相差不大,重复现象较好。此外,HT柔性纳米脂质体具有浅而宽的吸热峰,宽的熔区和较低的熔点表明它们具有有序性较低的晶体结构,有利于载药和包埋效率[23]。
2.2.4微观结构分析 利用透射电镜观察HT柔性纳米脂质体的微观结构,结果如图3所示。由图可知,整体上,HT柔性纳米脂质体的粒子具有不同形状,多呈圆形或者椭圆形,分布基本均匀,分散性较好。图3(c)中可明显看到HT纳米脂质体内部具有层状囊泡即同心圆的指纹状结构,其原因可能是胆酸钠的作用,使得脂质体双分子的变形性提高。

a.2 μm;b.500 nm;c.100 nm
基于已有研究[24]及本研究工艺优化及性能分析结果可知:HT柔性纳米脂质体将是一种有前途的、更高效的、更安全的递送系统,可用于保护生物活性羟基的损失,从而实现人体内的靶向递送,其可以在改善羟基酪醇的低化学稳定性、吸收性和生物利用度的前提下,更大程度地利用自身优势,透皮给药到所需的特定部位,避免肝脏首过效应,减少毒性。
2.3 贮藏稳定性结果分析
为了评价HT柔性纳米脂质体的贮藏稳定性,测定HT柔性纳米脂质体在不同温度和不同时间的包封率、粒径、PDI和Zeta电位,结果如图4所示。

a.包封率encapsulation efficiency; b.粒径grain size; c.PDI; d.Zeta电位Zeta potential图4 HT柔性纳米脂质体的贮藏稳定性
由图4(a)可知,随着贮藏时间的增加,包封率整体呈现下降趋势,且温度越高,HT柔性纳米脂质体包封率下降越快;在4 ℃条件下,贮藏28 d时包封率由46.78%下降到37.56%,下降趋势相对于25和60 ℃较为缓慢,原因可能是温度的升高,加速了脂质体膜材的分子热运动,使得脂质双分子层的酰基侧链从有序排列到无序,导致脂膜由凝胶状态变成液晶态,膜的横切面增加,双分子层的厚度减小,膜流动性增加,导致包埋的HT发生泄露[25]。
在图4(b)可知,随着贮藏时间的增加,粒径整体上呈现上升趋势,但温度越高粒径增加越快速;4 ℃下,贮藏28 d后,粒径从104.7 nm增加到152.7 nm,而60 ℃下的粒径则增加到了1 084.5 nm。原因可能是贮藏时脂质体体系受温度影响,导致磷脂膜之间发生了融合、聚集等现象,使其在用激光粒度仪检测时检测的是融合后的聚集体,显示粒径变大[26]。
PDI是聚合物分散性指数,用于描述聚合物相对分子质量分布,值越大,相对分子质量分布越宽,值越小,相对分子质量分布越均匀。由图4(c)可知,随着贮藏时间的增加,PDI整体上呈现增加趋势,温度越高,增加越明显,值越大,说明体系中脂质体分布越不均匀,原因可能也是贮藏时脂质体体系受温度影响,导致磷脂膜之间发生了融合、聚集等现象[26]。
Zeta电位是表征胶体分散系稳定性的重要指标,能够度量颗粒之间相互排斥或吸引的力的强度,粒子越小,Zeta电位的绝对值(正或负)越高,体系越稳定,即溶解或分散可以抵抗聚集的能力越强。一般情况,当Zeta电位的绝对值大于40 mV时,说明体系具有较好的稳定性,由图4(d)可知,随着贮藏时间的增加,Zeta电位绝对值整体上呈现下降趋势,温度越高,其绝对值越低,说明较高的温度极大破坏了体系的稳定性,原因可能也是贮藏时脂质体体系受温度影响,导致磷脂膜流动性增强,由凝胶状态变成液晶态,粒子表面电位发生改变,导致Zeta电位绝对值减小。
根据脂质体在4、 25和60 ℃贮藏28 d后的贮藏稳定性结果可知,该脂质体在4 ℃下贮藏各项指标均优于25和60 ℃,说明HT柔性纳米脂质体是一个热不稳定体系,低温比较有利于HT柔性纳米脂质体的稳定贮藏。
3 结 论
3.1利用薄膜分散法制备羟基酪醇(HT)柔性纳米脂质体,以其包封率为响应值,通过单因素试验、Plackett-Burman试验和Box-Behnken响应面优化试验,得到制备HT柔性纳米脂质体的较佳工艺条件为:HT用量1.5 mg、氢化卵磷脂与胆固醇质量比为3.3∶1、FA-F127用量为1 mg、胆酸钠用量为20.6 mg、超声波功率为10%(总功率650 W)、超声波作用时间为11 min,此时制备的HT柔性纳米脂质体平均包封率为46.78%。
3.2HT柔性纳米脂质体的结构表征结果显示:HT柔性纳米脂质体是有利于载药和包埋的,HT负载增强了空白脂质体的热稳定性,其粒子的微观结构多呈圆形或者椭圆形,分散性良好,且可见同心圆的指纹结构,将是一种有前途的更高效更安全的递送系统。
3.3较优工艺条件下制备的HT柔性纳米脂质体平均粒径为104.7 nm、PDI为0.231、Zeta电位为-42.5 mV。在4、 25和60 ℃分别贮藏28 d后发现:4 ℃比较有利于HT柔性纳米脂质体的稳定贮藏。
