反复横纹肌溶解、肾功能不全伴血色病一例
2023-09-28周梦兰叶文玲高瑞通
周梦兰,张 磊,叶文玲,高瑞通
中国医学科学院北京协和医院 1检验科 2肾内科,北京 100730
1 病例资料
患者男性,45岁,因“反复肌痛39年,肌酐升高3年余”于2022年2月就诊于北京协和医院肾内科。
患者于1983年(6岁)持续劳累后出现全身肌肉酸痛伴四肢肌无力,尿液呈酱油色,卧床休息后逐渐好转。此后患者于劳累或受凉后反复出现肌肉酸痛和肌无力症状,轻时仅累及双侧大腿,重时四肢均可受累,无法完成行走、蹲起、梳头等日常活动,需卧床休息至少24 h才可逐渐缓解;1984年至2021年11月期间累积发作8次,发作时间主要为冬季。2017年发作期间查尿常规示蛋白阳性(+),潜血阳性(+++),尿红细胞2个/μL(正常范围:0~8.4个/μL)。2018年8月发作期间查血肌酐(creatinine,Cr)为98 μmol/L(正常范围:57~97 μmol/L),此后监测Cr波动于101~110 μmol/L。
患者2013年体检时发现铁蛋白(ferritin,Fer)明显升高(>1000 μg/L,正常范围:24~336 μg/L)。2019年MRI示肝脏、脾脏铁沉积,心脏、肾脏未见异常;肝穿刺活检示肝实质及间质铁沉积,诊断为血色病。予规律放血治疗后,Fer由>2000 μg/L降至214 μg/L,转铁蛋白饱和度(transferrin saturation,TS)由72.8%降至25.1%(正常范围:25%~50%)。
2021年12月,患者受凉后再次出现四肢肌肉酸痛伴肌无力。分别于2021年12月30日、2022年1月25日就诊于外院和北京协和医院肾内科门诊,期间无特殊治疗,于北京协和医院查尿蛋白、尿潜血均为阴性,主要血生化指标变化见表1。
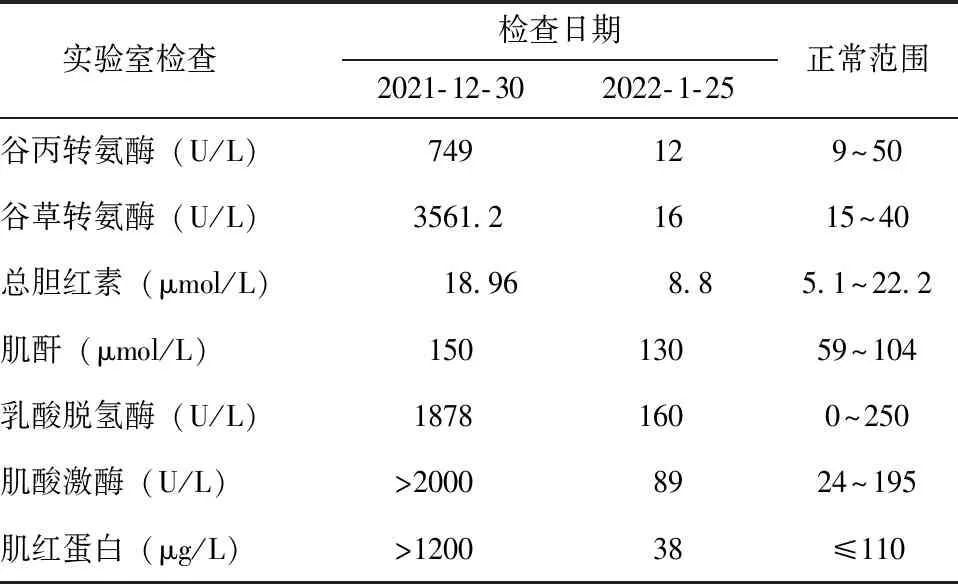
表 1 2021年12月发病后患者血生化指标变化
为进一步诊治,患者于2022年2月10日收住北京协和医院肾内科病房。起病以来,患者精神、饮食、睡眠均尚可,体质量无明显变化。家族史:1名兄长曾有寒冷、饥饿或劳累后四肢无力、酱油色尿病史,未特殊诊治,否认血色病家族史。
入室查体:心率78次/min,血压130/78 mm Hg(1 mm Hg=0.133 kPa),步行入室。双肺听诊呼吸音清,心律齐,心脏各瓣膜区未闻及杂音;腹软,无压痛及反跳痛;四肢肌容量、肌张力正常,肌力Ⅴ级。
患者中年男性,慢性病程,自幼起病。临床以发作性肌肉酸痛伴肌无力、酱油色尿、肾功能不全为主要表现,症状反复于劳累或受凉后出现。根据肌痛、肌无力等临床症状及肌酶升高等实验室检测结果可诊断为横纹肌溶解,其病因排除外伤、挤压伤、热射病、癫痫发作、低钾血症、多发性肌炎、镰状细胞性贫血等,鉴别诊断主要集中于代谢性肌病和线粒体肌病,可进一步完善血脂酰肉碱谱、肌电图、基因检测等,并请神经科、儿科协助诊治。肾脏方面主要表现为慢性肾功能不全,肌病发作时Cr可急性升高,由于无明显血尿、蛋白尿,考虑病变主要定位于肾小管间质,现阶段需重点考虑以下病因:(1)横纹肌溶解急性发作时可导致肾内血管收缩、肾小管损伤,肌红蛋白管型阻塞肾小管时亦可引起急性肾损伤,反复发作可形成慢性肾小管间质病变;(2)患者于2019年诊断为血色病,伴肝脏、脾脏铁沉积,虽然肾脏非血色病的常见铁沉积部位,但有血色病引起肾脏铁沉积的个案报道[1],可予以肾脏穿刺病理检查并行特殊铁染色。
入院后完善相关检查:血常规示白细胞4.49×109/L,血红蛋白122 g/L,血小板200×109/L;24 h尿蛋白0.09 g;肝肾功能示谷丙转氨酶14 U/L,谷草转氨酶23 U/L,总胆红素14.2 μmol/L,Cr 120 μmol/L,电解质、酸碱平衡均正常;Fer 237 μg/L,TS 26.5%;肌酸激酶 297 U/L,肌红蛋白 190 μg/L,心肌肌钙蛋白Ⅰ<0.017 μg/L;抗核抗体谱、血清蛋白电泳、冷球蛋白均为阴性。横纹肌溶解病因筛查方面,儿科、神经科会诊后考虑患者幼年起病,有家族史,代谢性肌病可能性大。线粒体病较罕见,予进一步完善检查以辅助诊断,结果示血乳酸0.8 mmol/L,血氨20 μmol/L,均正常。血脂酰肉碱谱检测发现游离肉碱(C0)为11.489 μmol/L,十四烯酰肉碱(C14∶1)为0.082 μmol/L,十二烯酰肉碱(C12∶1)为0.035 μmol/L,均正常;十四碳二烯酰肉碱(C14∶2)为0.155 μmol/L(正常范围:0~0.13 μmol/L),十四酰肉碱(C14)为0.176 μmol/L(正常范围:0~0.12 μmol/L),均升高。肌电图未见神经源性及肌源性损害。全外显子组测序发现极长链酰基辅酶A脱氢酶(very long chain acyl-CoA dehydrogenase,VLCAD)基因复合杂合突变(c.1246G>A和c.664G>C),提示为VLCAD缺乏症(OMIM:201475);未检出与临床表型相关线粒体基因突变和血色病相关基因突变。
为明确肾功能不全的病因,予完善肾穿刺活检,结果示免疫荧光阴性。光镜下肾小球无明显病变;可见肾小管刷状缘脱落和上皮细胞扁平化以及肾小管上皮细胞修复再生现象;可见数处灶性至小灶性分布的肾小管萎缩;间质可见数处灶性至小灶性分布的纤维化;未见脂质沉积(图1)。进一步行普鲁士蓝染色未见肾脏铁沉积。综合考虑诊断为肾小管间质损伤,急慢性病变均可见。
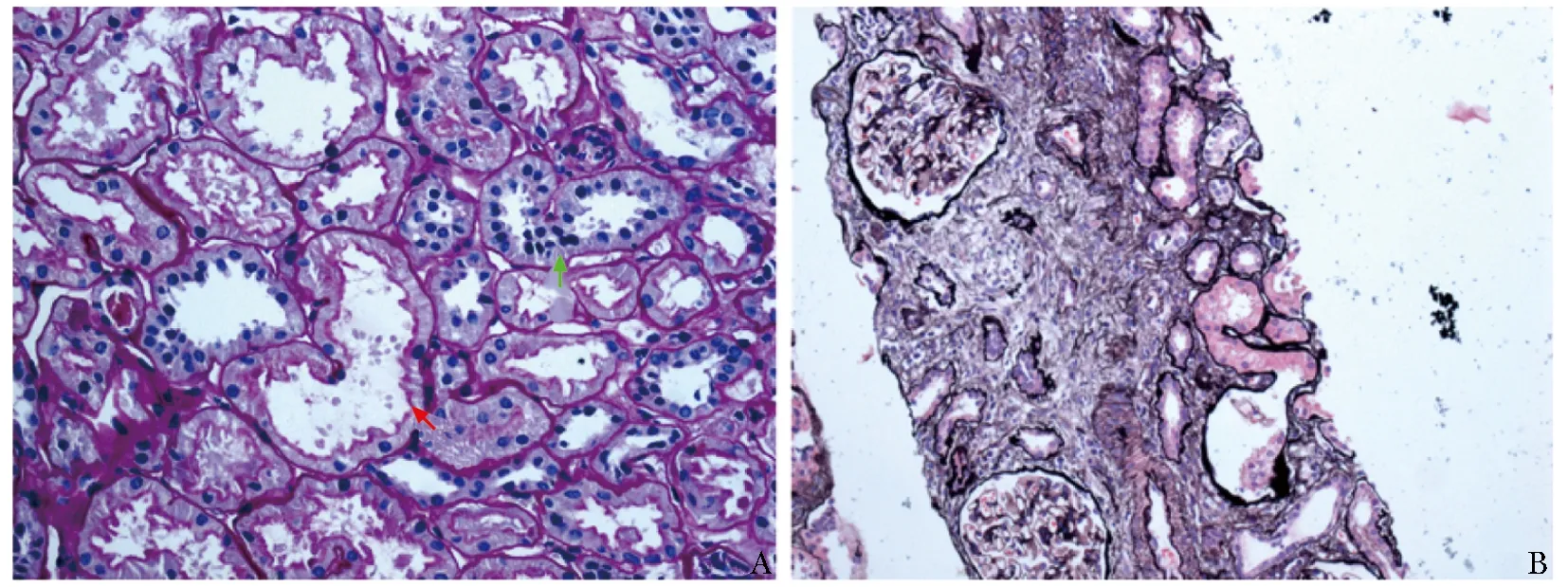
图 1 患者肾脏穿刺病理表现
综上,患者反复肌痛、肌无力伴Cr升高,具有血色病的背景。辅助检查示部分血脂酰肉碱浓度升高;肾脏穿刺活检可见肾小管间质损伤,未见脂质及铁沉积;全外显子组测序示ACADVL基因复合杂合突变,综合考虑后3型VLCAD缺乏症诊断明确,肾功能不全为该病所致横纹肌溶解的并发症。
嘱患者自主多饮水(2500~3000 mL/d),未再出现肌肉酸痛、酱油色尿等不适症状,顺利出院。出院后患者一般状况可,嘱日常生活中尽量避免劳累、受凉、饥饿、感染等诱因,未再出现横纹肌溶解发作。末次随访(2022年11月)时尿蛋白、尿潜血均为阴性,血Cr 117 μmol/L,肌酸激酶133 U/L,Fer 121 μg/L,TS 27.5%。
VLCAD缺乏症是由于ACADVL基因突变以致细胞线粒体内脂肪酸β氧化过程中的关键酶VLCAD缺乏所致的常染色体隐性遗传病。VLCAD缺乏将导致体内长链脂肪酸代谢障碍、长链脂肪酸贮积,进而对心肌、骨骼肌、肝脏等产生毒性作用[2]。既往认为VLCAD缺乏症发病率约为1/120 000~1/100 000,但来自亚洲、欧洲、美洲多个地区的新生儿筛查数据显示其发病率可达1/42 500[3],由于临床较罕见,该病已被收录至2018年国家卫生健康委员会联合多部门制定的《第一批罕见病目录》中[4]。根据起病年龄和临床表现,可将VLCAD缺乏症分为3个亚型:严重早发心脏和多脏器衰竭型、肝脏和低酮性低血糖型、迟发性间歇肌病型,此外亦有无症状型的报道[5]。迟发性间歇肌病型VLCAD缺乏症最为常见,青少年或成年发病,表现为运动、感染、饥饿等诱发的横纹肌溶解和肌红蛋白尿[2,6]。该病诊断主要依靠临床表现、生化检测和基因检测。对临床疑诊VLCAD缺乏症的患者,均应行血脂酰肉碱谱分析,发现C14∶1>1000 μmol/L强烈提示本病,若进一步ACADVL基因检测可见2个等位基因致病突变可确诊[2]。本例患者具有3型VLCAD缺乏症的典型临床表现,结合基因检测结果可明确诊断。VLCAD缺乏者最主要的代谢表现包括C14∶1、C14∶2、C14及C12∶1浓度升高,其中以C14∶1升高最为明显[7-8]。该患者血脂酰肉碱谱检测结果不典型,考虑与检测时处于非应激期或病情稳定期相关[5,8]。
肾脏病理对肾功能不全的病因鉴别具有重要意义。分析本例患者肾功能不全的可能原因,首先考虑与VLCAD缺乏症引起横纹肌溶解相关[5,9]。急性肾损伤是横纹肌溶解的严重并发症之一,文献报道,横纹肌溶解患者发生急性肾损伤的比例高达13%~50%,此类急性肾损伤占所有急性肾损伤的7%~10%[10]。急性肾损伤的发生机制主要包括肾内血管收缩、肾小管损伤、肾小管阻塞以及补体活化[10-11]。肾脏病理可见急性肾小管坏死或急性肾小管损伤、肌红蛋白管型、肾间质水肿伴单核细胞浸润[12-13],而反复发生横纹肌溶解可形成慢性肾小管间质病变。研究显示,若入院时肌酸激酶水平低于15 000~20 000 U/L,则继发急性肾损伤的风险较低,而低肌酸激酶患者发生急性肾损伤的危险因素包括脱水、酸中毒和脓毒血症[10]。本例患者2021年12月30日发作期间测定的肌酸激酶高于检测值上限,推测可能导致急性肾损伤的风险较高。2022年2月10日入院后肾脏病理既可见急性肾小管损伤表现,亦具有慢性肾小管萎缩、间质纤维化的表现,与其病史特点较为相符。
遗传性血色病(hereditary hemochromatosis,HH)最常由HFE基因的C282Y纯合突变所引起,可导致铁吸收增加,铁沉积的主要器官为肝脏和关节[14],但其他部位亦可受累。研究显示,HH患者多在40~50岁时被诊断,肝脏、胰腺、关节、心脏、皮肤和垂体为常见的受累器官[15]。该患者的血色病原因尚不明确,未发现与VLCAD缺乏症有共同致病突变,故推测可能为临床合并症,但血色病亦可引起肾脏铁沉积。个案报道显示,血色病引起的肾脏铁沉积主要部位为肾小管,并可引起先天性近端肾小管发育不良[16-17]。Nakayama等[1]报道了1例55岁男性血色病患者,其24 h尿蛋白为1.12 g,可见镜下血尿、肾功能不全(Cr 55 mg/L);肾活检示IgA肾病合并肾脏铁沉积,主要沉积部位为肾小球及肾小管上皮细胞。Ozkurt等[18]报道了1例61岁男性血色病患者发生急进性肾小球肾炎的病例,肾活检可见肾小球新月体形成,肾小管上皮细胞内广泛含铁血黄素沉积。本例患者MRI及普鲁士蓝染色均未发现肾脏铁沉积的证据,考虑其肾功能不全与血色病无明确相关性。
此外,还需鉴别肾脏疾病是否与VLCAD缺乏症脂质沉积相关。尽管3型VLCAD缺乏症以骨骼肌受累为主,但亦有3型VLCAD缺乏症引起心肌病、呼吸衰竭、低血糖等非骨骼肌症状的报道,提示该病可能导致肌病以外的症状[5]。目前,VLCAD缺乏症相关肾脏脂质沉积的文献较匮乏,Hasegawa等[19]报道了1例VLCAD缺乏症男婴,患儿出生118 d时因室颤去世,尸检示肝脏、心肌、肾脏、骨骼肌和肠黏膜中大量脂质沉积。本例患者肾组织中并未见脂质沉积的证据,亦排除VLCAD缺乏症脂质沉积引起的肾损伤。
VLCAD缺乏症的总体治疗原则为避免空腹以及急性发作的诱因,合理饮食,对症干预,控制急性发作并积极治疗并发症。在3个亚型中,以严重早发心脏和多脏器衰竭型最为凶险,病死率高,即使早期诊断和干预,总体预后仍较差[5,8]。本例患者针对肾功能不全进行了积极水化处理,并嘱患者日常生活中尽量避免劳累、受凉、饥饿、感染等诱因,未再出现横纹肌溶解发作。
综上,本文首次报道了1例3型VLCAD缺乏症合并血色病的罕见病例,其肾脏病变因VLCAD缺乏症引起的反复横纹肌溶解所致,表现为慢性肾小管间质损伤,且在急性发作时合并急性肾小管损伤。因患者临床合并症较多,对肾功能不全的病因鉴别需警惕有无肾脏铁沉积以及脂质沉积可能,而肾脏病理穿刺活检可为肾脏损伤病因的鉴别提供重要参考信息,尤其对于伴复杂基础疾病的患者,肾脏病理穿刺除可提供常规信息外,还可通过多种特殊染色为罕见病因提供鉴别诊断思路。
作者贡献:周梦兰、张磊负责论文撰写及修订;叶文玲、高瑞通负责提出修改意见。
利益冲突:所有作者均声明不存在利益冲突
注:本研究发表已获得患者知情同意。
