BHA 浮选锡石体系中锌组分活化机理的量子化学计算研究
2023-09-19代龙富郝佳美秦晓艳高虎林
代龙富 刘 建,2 李 达 郝佳美 秦晓艳 高虎林
(1.昆明理工大学国土资源工程学院,云南 昆明 650093;2.省部共建复杂有色金属资源清洁利用国家重点实验室,云南 昆明 650093)
锡是人类历史上最早被利用的金属之一,同时也是我国重要的战略金属之一[1-2]。锡具有无毒、熔点低、耐腐蚀、质地软、延展性好等优良性质,被广泛应用于电子、化工、冶金、机械、食品包装、原子能及航天工业等领域[3-4]。锡石中理论锡含量为78.8%,是地壳中最常见的含锡矿物,且被认为是唯一可利用的锡矿物。锡石由四方结构的二氧化锡组成,具有高密度、强硬度和高脆性,使其适用于重选分离[5-7]。但随着高品质锡石的逐渐枯竭,低品位细粒锡石和重选尾矿正在成为锡石浮选原料的主要来源;由于锡石浮选捕收剂的发展及应用,使得这类锡石的有效回收成为可能[8-11]。量子化学计算是研究矿物的晶体结构、电子性质及表面吸附的有效方法,在矿物表面吸附化学等领域有着非常重要的应用[12-16]。根据密度泛函理论可获得矿物表面的微观结构及物理化学信息,如通过前线轨道分析可以判断得失电子中心、HOMO 与LUMO 分布情况、轨道组成等信息;利用电荷布居分析可以获得各原子电荷差异,进而判断物质的反应活性中心;利用反应物与生成物最优结构的能量,计算得到相互作用能,进一步判断反应物之间作用能力强弱[17-21]。这为研究锌组分在锡石的BHA 浮选体系中的作用机理提供了有力的帮助。
在锡石浮选中,金属离子有着非常重要的作用,其中铅离子被广泛应用于羟肟酸类浮选锡石,但其具有很强的生物毒性,难以进行生物化学降解,从而导致生物化学和环境问题。因此,有学者已经在寻找用于锡石浮选的替代金属离子[22-30]。锌的生物毒性和环境影响远小于铅,研究锌组分是否具有在羟肟酸类捕收剂存在下活化锡石的潜力是十分必要的。CAO[31]研究了锌组分在BHA 浮选锡石体系中的活化作用,结果表明,锌组分的存在增加了BHA 在锡石表面的吸附量,且锌组分的活化效果高于铅组分,锡石最大的浮选回收率可达90.54%。迄今为止并没有从原子微观角度对锌组分活化锡石表面的作用机理进行系统详细的探究,BHA 浮选体系中锌组分活化锡石表面的量子化学研究工作亟须进行。
为了揭示锌组分在BHA 浮选锡石体系中的活化机理,本研究基于密度泛函理论,建立了锡石表面上锌组分和BHA 的共吸附模型,并采用第一性原理计算方法研究了锌组分在BHA 浮选锡石体系中的作用机理,从原子微观角度定性研究了锌组分吸附于锡石表面后的电子性质、电荷转移情况及吸附性能,为锌组分活化锡石浮选的实践提供重要的理论指导。
1 模型建立及计算方法
1.1 计算方法
采用Material Studio 中CASTAP 模块进行所有的结构优化、能量计算和性质分析。根据前人经验[11,32-34],计算中交换关联函数采用广义梯度近似(GGA)下的PBE 梯度修正函数,采用Customized 赝势描述离子核和价电子之间的相互作用,价电子平面波函数截断能(Energy cutoff)设置为450 eV,所有计算均在倒易空间中完成,k 点(k-point)网络设置为2×2×1,自洽场收敛精度设置为2.0×10-6eV/atom;原子间相互作用力(Max.force)收敛标准设置为0.01 eV/Å(1 Å=0.1 nm);晶体内应力的收敛标准(Max.stress)设置为0.2 GPa;最大循环(Max.iterations)设置为500。由于锡石是非磁性矿物,后续所有计算过程中均采用非自旋进行计算。
吸附能是一个或多个分子在吸附界面上方运动时,在其速率由大变小并最终吸附在吸附界面上的过程中释放出的能量。不同分子或离子与相同矿物表面作用时的吸附能有差异,由此可凭借吸附能来判断药剂与矿物表面的吸附作用,吸附能为负值时说明吸附可以自发进行,吸附能的值越小代表着吸附越稳定;吸附能为0 或正值时表明吸附不能自发进行。药剂分子在矿物表面上的吸附能计算如下式所示:
ΔE=Ecom-Esurf-Erea,
式中:ΔE为吸附能,Ecom为吸附络合物的总能量,Esurf为表面结构的总能量,Erea为吸附质的总能量,单位均为kJ/mol。
1.2 锡石晶体及表面的构建及优化
锡石化学式为SnO2,属于四方晶系的氧化矿物,具有金红石构型,通常为双锥状或细长柱状的短柱体[5-6]。通过几何优化得到的锡石晶体单胞的晶格参数如表1 所示,与实验值相对误差均小于0.01%,可进行后续计算。
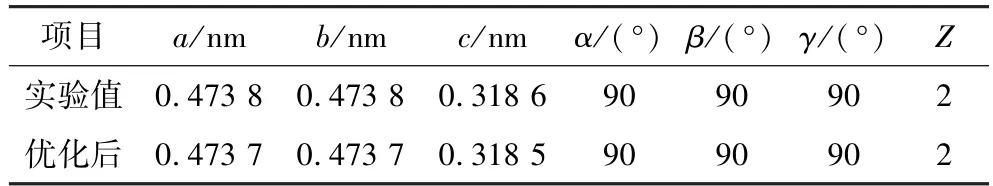
表1 经几何优化后锡石晶体单胞模型的晶格参数Table 1 Lattice parameters of cassiterite crystal unit cell model after geometric optimization
锡石作为典型的AX2型氧化矿物,晶体结构是六方最紧密堆积,其表面疏水性较弱。通常,在外力的作用下,矿物一般会沿着平行于层间距较大的面、阴阳离子电性中和的面、两层离子相邻的面和化学键合强度最弱的方向产生破碎。研究表明(110)表面的表面能与断键密度远小于(101)面和(111)面,是锡石表面暴露最多的表面,可将(110)表面作为锡石颗粒的代表性表面[32]。因此,本研究均在锡石的(110)表面上进行。考虑到后续锌组分与BHA 分子在锡石表面的吸附,采用2×2×2 的超胞模型,设置真空层为1.5 nm,共3 个原子层,共包含24 个Sn 原子和48 个O 原子共72 个原子;为提高计算效率,固定底部两层原子。
图1 为经几何优化后的锡石(110)面模型,锡石(110)面呈现出1 个双配位的O(顶位)原子与2 个六配位的Sn 原子结合在1 个平面上,和1 个三配位的O(底位)原子与2 个五配位的Sn 原子和1 个六配位的Sn 原子相连接,整体呈一个类似凸凹的结构。
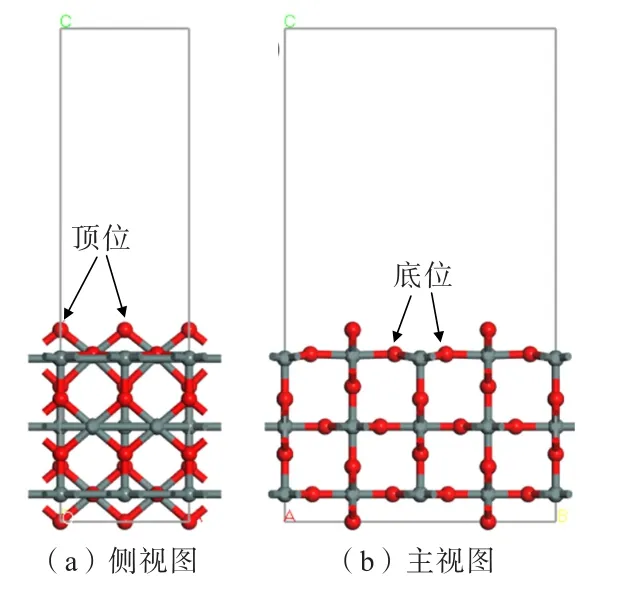
图1 经几何优化后的锡石(110)面模型Fig.1 Geometrically optimized cassiterite (110)surface model
1.3 药剂分子模型的构建及优化
CAO 等[31]研究结果表明:以BHA 和ZnSO4浮选锡石时,最佳pH 条件为7.5 ~8.0。由溶液化学计算结果可知,溶液中的锌组分在pH 为7.5~8 时主要以游离的Zn(OH)+的形式存在[31,35]。溶液中的BHA在pH=7.5~8 时主要以游离的BHA 分子的形式存在(平衡浓度:pKa=8.5 ~9.0)[31,36]。因此,本文以Zn(OH)+代表锌组分与BHA 分子进行计算。在3D模型中构建Zn(OH)+分子与BHA 分子结构模型,再采用CASTAP 模块对Zn(OH)+分子与BHA 分子的结构模型分别进行结构优化,计算中交换关联函数采用广义梯度近似(GGA)下的PBE 梯度修正函数,采用Fine 赝势描述离子核和价电子之间的相互作用。优化后的Zn(OH)+分子与BHA 分子的结构如图2所示。
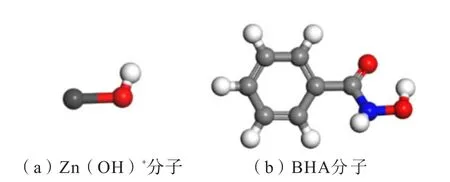
图2 经几何优化后的药剂分子结构模型Fig.2 Molecular structure model of reagent after geometric optimization
2 试验结果与讨论
2.1 锡石(110)面的态密度分析
首先计算了未吸附药剂分子前的锡石(110)面各原子分态密度及总体态密度,结果如图3 所示。
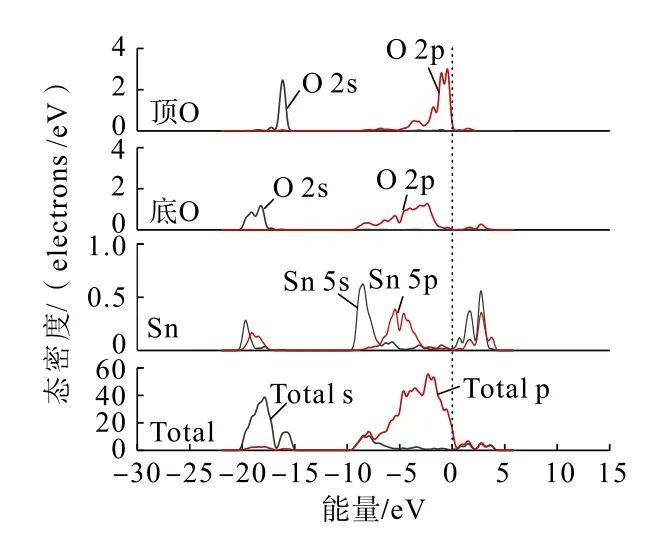
图3 锡石(110)面各原子分态密度及总体态密度Fig.3 Partial and total density of states of each atom on the surface of cassiterite (110)
通过锡石(110)解离面的态密度分析可以发现:在-20~-15 eV 价带主要由O(顶位)原子与O(底位)原子的2s 轨道以及少量Sn 的5s、5p 轨道组成;在-9~1 eV 价带主要由O(顶位)原子与O(底位)原子的2p 轨道以及部分Sn 的5s、5p 轨道组成;在1 ~4 eV价带主要由Sn 的5s、5p 轨道。在费米能级附近主要由O(顶位)原子的2p 轨道组成,说明O(顶位)原子的活性较强,容易参与反应。
2.2 锌组分在锡石表面上的吸附情况
在矿物浮选中,药剂分子在矿物表面上的吸附取决于吸附位点及其活性与吸附形式,2.1 节已说明锡石(110)表面的O(顶位)原子活性较强。故本节根据Zn(OH)+在锡石(110)面O(顶位)原子位点上不同的吸附形式,分别计算了Zn(OH)+在锡石(110)面O(顶位)原子位点上单桥形式与双桥形式的吸附情况。
2.2.1 吸附能
图4 为Zn(OH)+在锡石(110)面O 原子位点上不同形式的吸附模型。单桥形式吸附时,Zn(OH)+在锡石表面1 个O(顶位)原子斜上方,与1 个O(顶位)原子形成线吸附;双桥形式吸附时,Zn(OH)+吸附在锡石(110)面的2 个O(顶位)原子上,形成面吸附。计算得到的吸附能如表2 所示。
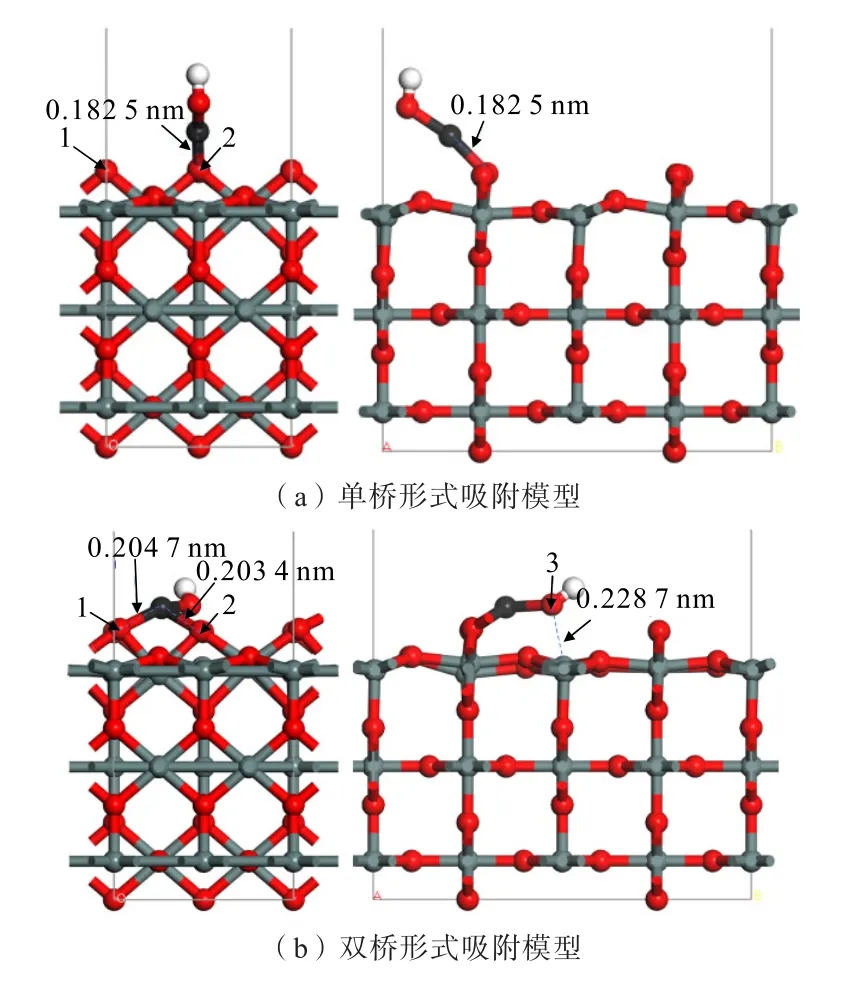
图4 Zn(OH)+在锡石(110)面上不同形式的吸附模型Fig.4 Different adsorption models of Zn(OH)+on cassiterite (110) surface

表2 Zn(OH)+在锡石(110)面上不同吸附形式的吸附能及键长Table 2 Different adsorption forms of adsorption energy and bond length of Zn(OH)+on cassiterite (110) surface
从表2 可知,Zn(OH)+在锡石(110)表面上单桥与双桥形式吸附时的吸附能分别为-532.39、-580.64 kJ/mol,吸附能均为负值,说明Zn(OH)+可在锡石(110)面上自发吸附。
由图4 可知,单桥形式吸附的最佳吸附构型中,Zn(OH)+中Zn 原子与锡石表面1 个O(顶位)原子形成的Zn—O 键长为0.182 5 nm,小于Zn、O 原子的半径之和(0.205 nm),说明了Zn(OH)+在锡石表面上的O(顶位)原子发生了化学吸附。双桥形式吸附的最佳吸附构型中,Zn—O 键长分别为0.204 7、0.203 4 nm;Zn(OH)+中Zn 原子与两个O(顶位)原子生成的Zn—O 键长小于Zn、O 原子的半径之和(0.205 nm),发生了化学吸附;此外,Zn(OH)+中的O原子向锡石(110)面上的Sn 原子处发生偏移,原子间距为0.228 7 nm,大于Sn、O 原子的半径之和(0.224 0 nm),说明了Zn(OH)+中的O 原子与锡石(110)面上的Sn 原子未发生化学吸附,未形成稳定的Sn—O 键。
总体比较Zn(OH)+分别与锡石(110)面上不同形式的吸附情况,发现Zn(OH)+均能以不同形式与锡石(110)面发生吸附。其中,双桥形式的吸附能更负,吸附作用更稳定,且Zn(OH)+中O 原子向锡石(110)面上的Sn 原子处发生偏移,形成了更稳定的面吸附形式。说明Zn(OH)+在锡石(110)面上的吸附主要是发生面吸附的双桥形式。
2.2.2 态密度
为了从原子角度直观分析Zn(OH)+在锡石(110)面上不同形式的吸附情况,利用态密度分析Zn(OH)+在锡石(110)面O 原子(图中O 原子序号见图4)位点上以不同形式吸附前后,各轨道组成的贡献情况。吸附前后的锡石(110)面上O 原子与Zn原子的态密度分别如图5、图6 和图7 所示。
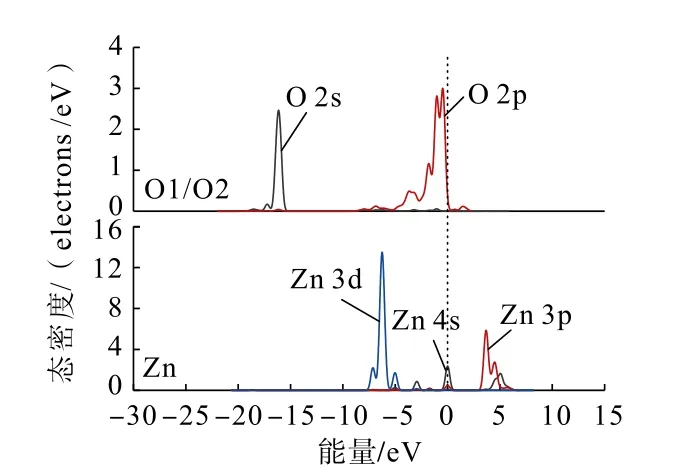
图5 吸附前锡石(110)面上O 原子与Zn(OH)+中Zn 原子的分态密度Fig.5 Partial density of states between O atoms on cassiterite (110) surface and Zn atoms in Zn(OH)+before adsorption
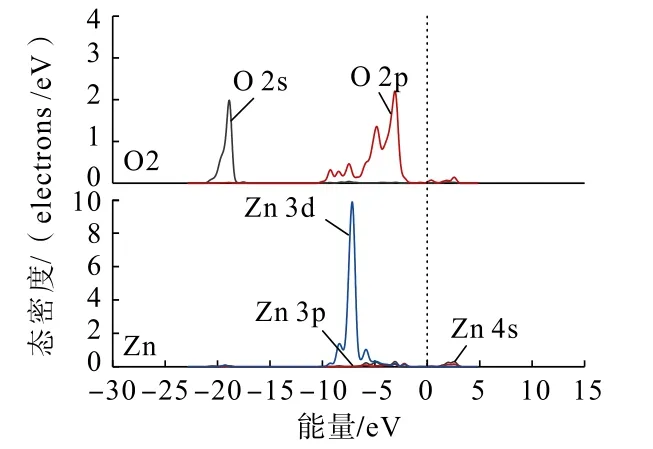
图6 单桥形式吸附后锡石(110)面上O 原子与Zn(OH)+中Zn 原子的分态密度Fig.6 Partial density of states between O atoms on cassiterite (110) surface and Zn atoms in Zn(OH)+after single bridge adsorption
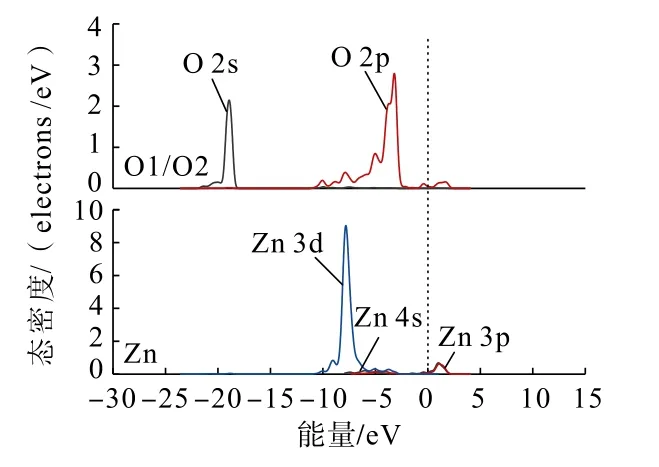
图7 双桥形式吸附后锡石(110)面上O 原子与Zn(OH)+中Zn 原子的分态密度Fig.7 Partial density of states between O atoms on cassiterite (110) surface and Zn atoms in Zn(OH)+after double bridge adsorption
通过对比Zn(OH)+在锡石(110)面O(顶位)原子位点上以不同形式吸附前后的态密度可以发现:Zn(OH)+以不同形式吸附后,锡石表面O 原子的2s、2p 轨道与Zn 原子的3 d 轨道均向低能量方向分别移动,说明在加入Zn(OH)+后Zn、O 原子经成键相互作用生成Zn—O 键,使得Zn(OH)+能够吸附在锡石(110)面。双桥形式吸附时,Zn 原子的3d 轨道较双桥形式吸附时向低能量方向移动更深,说明了Zn(OH)+以双桥形式吸附在锡石(110)面较为稳定。
Zn(OH)+以双桥形式吸附在锡石(110)面时,Zn(OH)+中O 会向锡石(110)面上的Sn 原子发生偏移。Zn(OH)+在锡石(110)面O 原子位点上发生双桥形式的吸附前后,对Zn(OH)+中O 原子与锡石表面Sn 原子进行态密度分析,态密度分别如图8 与图9 所示。
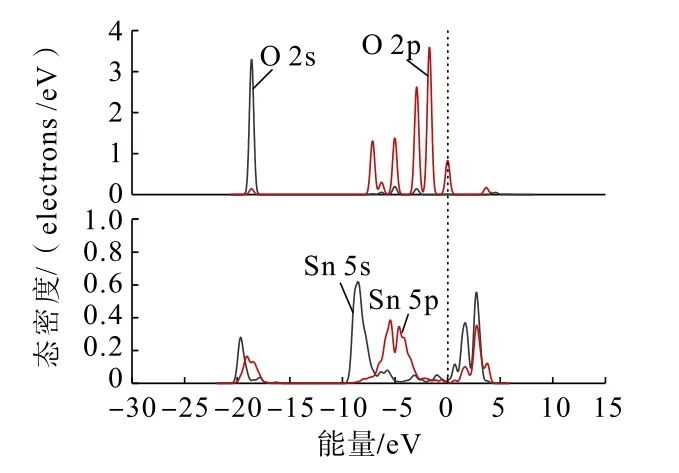
图8 Zn(OH)+中O 原子与锡石(110)面Sn 原子的分态密度Fig.8 Partial density of states between O atoms in Zn(OH)+and Sn atoms on cassiterite (110) surface
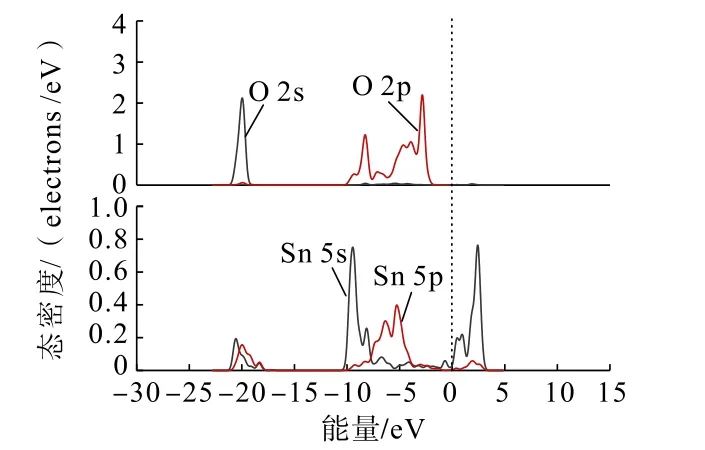
图9 双桥形式吸附后Zn(OH)+中O 原子与锡石(110)面Sn 原子的分态密度Fig.9 Partial density of states between O atoms in Zn(OH)+and Sn atoms on cassiterite (110) surface after double bridge adsorption
通过对比Zn(OH)+在锡石(110)面O 原子位点上以双桥形式吸附前后的态密度可以发现:Zn(OH)+吸附后,Zn(OH)+中O 原子的2s、2p 轨道与锡石表面Sn 原子的5s、5p 轨道均向低能量方向移动不明显,在费米能级附近的O 原子的2p 轨道峰数减少,越过费米能级在-4 ~-10 eV 处与Sn 原子的5s、5p 轨道未发生交叠,说明了在加入锌组分后,Zn(OH)+中O 原子未吸附在锡石表面的Sn 原子上,未形成Sn—O 键。这与Zn(OH)+在锡石(110)面上的吸附结果一致。
2.2.3 Mulliken 布居分析
在分子作用体系中,为了研究各原子的电荷分布、转移以及成键特性等性质,常通过Mulliken 布居进行分析判断。通过对Zn(OH)+以不同形式吸附在锡石(110)面上O 原子前后模型进行计算,得到了Mulliken 键布居与键长,如表3 所示(O 原子对应序号位置见图4)。
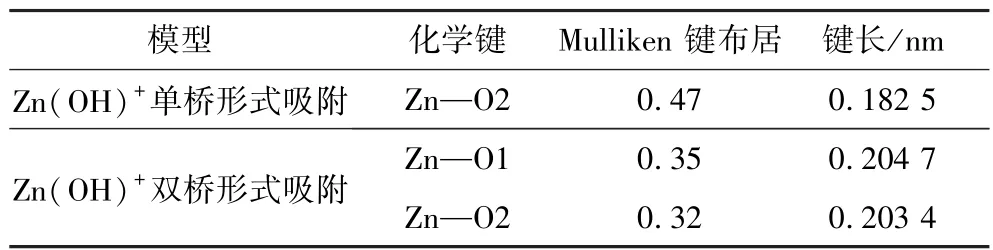
表3 Zn(OH)+以不同吸附形式吸附在锡石(110)面的Mulliken 键布居与键长Ta ble 3 Mulliken bond population and bond length of Zn(OH)+ adsorbed on cassiterite(110) surface in different adsorption forms
从表3 可以看出,Zn(OH)+以单桥形式吸附后,锡石(110)表面形成的Zn—O2 化学键的Mulliken 键布居和键长分别为0.32、0.182 5 nm,显然在Zn(OH)+以单桥形式吸附锡石(110)表面的过程中形成了稳定的共价键,可以认为Zn(OH)+以单桥形式与锡石表面的O2原子发生吸附,形成直线吸附结构。Zn(OH)+以双桥形式吸附后,锡石(110)表面形成的Zn—O1、Zn—O2 化学键的Mulliken 键布居和键长分别为0.35、0.204 7 nm 与0.32、0.203 4 nm,其中Zn—O1 与Zn—O2 两者的Mulliken 键布居和键长较为接近,成键稳定性好,各化学键的共价性差异较小,可以认为Zn(OH)+以双桥形式吸附在锡石(110)面时,受空间因素影响,与O1、O2 原子同时吸附,形成更为稳定的面螯合结构。
表4 为Zn(OH)+以不同吸附形式吸附在锡石(110)面各原子的Mulliken 电荷布居(O 原子对应序号位置见图4)。
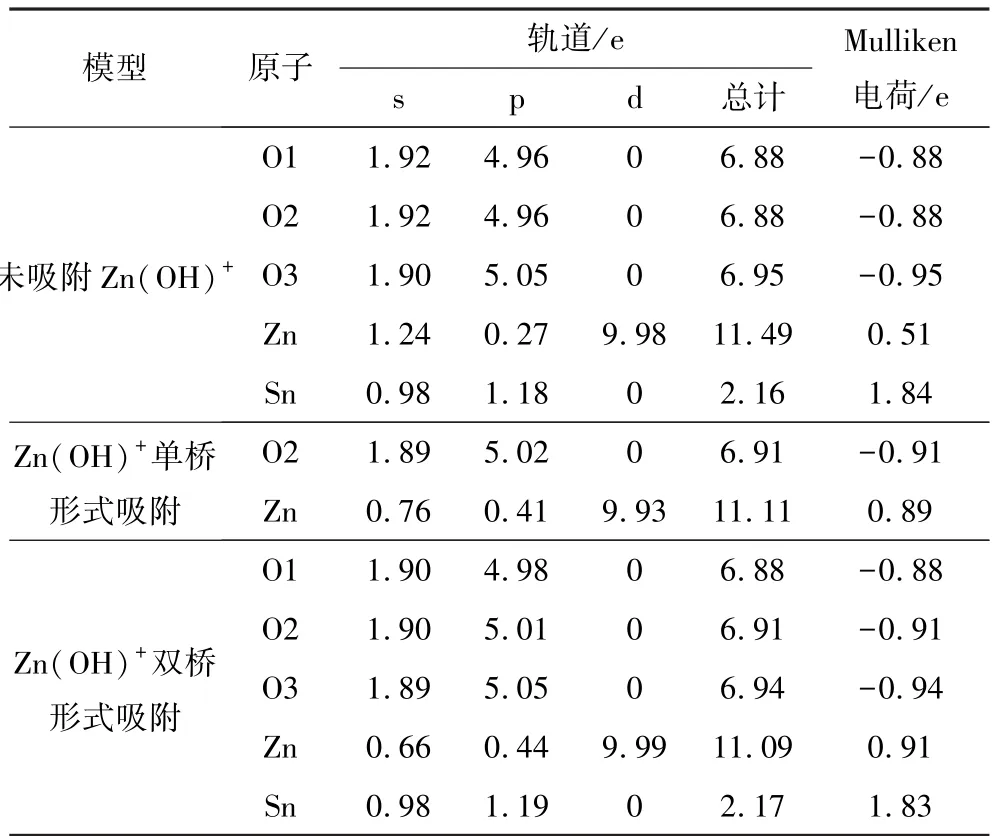
表4 Zn(OH)+以不同吸附形式吸附在锡石(110)面各原子的Mulliken 电荷布居Table 4 Mulliken charge population of Zn(OH)+adsorbed on each atom of cassiterite (110) surface in different adsorption forms
由表4 可知,Zn(OH)+以不同形式吸附在锡石(110)面O 原子位点上后,Zn 原子电荷总量均降低,其中4s 和3p 轨道的电荷发生了较大的变化,3d 轨道的电荷几乎无任何变化,结合Zn(OH)+吸附前后Zn 原子的态密度结果进行分析,在吸附过程中,Zn原子发生电荷转移可能是因为3d 电子转移后,4s 或3p 再次向3d 转移。同时,O 原子电荷总量升高,其2s、2p 轨道在吸附前后均发生了变化。在双桥形式吸附时,Sn 原子电荷总量与Sn 原子发生直接作用的O3 原子电荷总量均变化不明显,说明在吸附过程中,Sn 原子与Zn(OH)+中O3 原子未发生化学吸附。比较两种不同吸附形式的吸附模型可知,双桥形式吸附后各原子的转移电荷总数要大于单桥形式吸附后各原子的转移电荷总数,说明了Zn(OH)+吸附锡石时双桥形式吸附更稳定,这与吸附能和态密度的讨论结果相一致。
2.2.4 差分电荷密度分析
通过态密度及Mulliken 布居分析发现:锌组分在锡石(110)表面O 原子位点上吸附时,O、Zn 原子之间发生了电荷转移并形成了新的Zn—O 键,为了直观地分析电子的分布密度并判断原子之间的成键情况,对Zn(OH)+以不同形式吸附锡石(110)面后模型进行了差分电荷密度分析,结果如图10 所示。当两原子之间最低电荷密度颜色比背景板的颜色更深时,说明两原子之间成共价键,反之,成离子键;同时也可通过颜色的空间分布判断原子之间得失电子的情况。
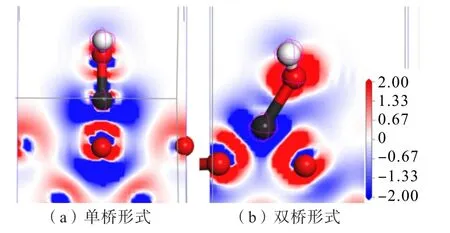
图10 Zn(OH)+以不同形式吸附锡石(110)面后的差分电荷密度Fig.10 Differential charge density diagram of Zn(OH)+adsorbed cassiterite (110) surface in different forms
通过差分密度图可知:Zn(OH)+在锡石(110)面的O 位点以不同形式吸附后,Zn 原子与锡石(110)面的O(顶位)原子之间均出现了电荷交互,说明Zn、O(顶位)原子之间发生了电子的转移,生成了Zn—O共价键;又由于Zn(OH)+之间原本就存在的化学键,因此也出现了电荷交互的作用。对比图10(a)与图(b)可以发现,Zn(OH)+在锡石(110)面的O 位点以双桥形式吸附时,Zn、O 原子之间最低电荷密度颜色比单桥形式的颜色更深,转移电荷量更多,说明Zn(OH)+吸附锡石时以双桥形式吸附更稳定,与前文讨论的结果相一致。
2.3 BHA 在锌组分作用后的锡石表面上吸附
由2.2 可知,Zn(OH)+以双桥形式吸附在锡石(110)面上吸附能较低,吸附稳定性好,故本节分别计算了BHA 与锡石(110)面、BHA 与Zn(OH)+以双桥形式作用后的锡石(110)面的吸附情况,讨论Zn(OH)+的加入对BHA 浮选锡石体系的作用机制。优化后的吸附模型如图11 所示,吸附能计算结果如表5 所示。
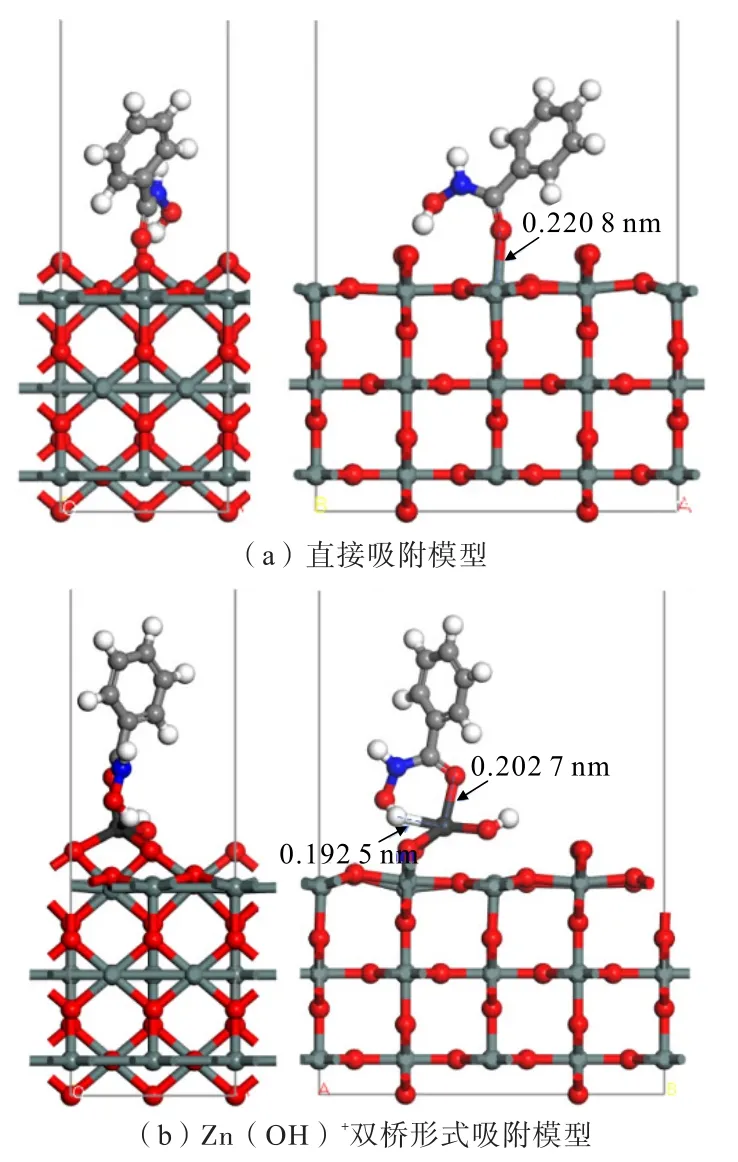
图11 Zn(OH)+加入前后,BHA 吸附于锡石(110)面的吸附情况Fig.11 The adsorption of BHA on cassiterite (110) surface before or after the addition of Zn(OH)+

表5 Zn(OH)+加入前后,BHA 吸附于锡石(110)面的吸附能Table 5 Adsorption energy of BHA adsorbed on cassiterite (110) surface before or after the addition of Zn(OH)+
表5 表明,BHA 与锡石(110)面直接吸附时,吸附能为-221.23 kJ/mol,吸附能为负,说明BHA 可自发地在锡石表面吸附;而加入Zn(OH)+对锡石(110)面作用后,BHA 与作用后的锡石(110)面吸附能为780.97 kJ/mol,为负值,且较BHA 与锡石(110)面直接吸附的吸附能更负,说明Zn(OH)+的存在有利于BHA 在锡石表面的吸附。如图11 所示的吸附模型,BHA 与锡石(110)面直接吸附时,BHA 分子中双键O原子与锡石(110)面的Sn 原子直接形成单键吸附;而加入Zn(OH)+对锡石(110)面作用后,BHA 分子中双键O 原子和—OH 基团与锡石(110)面上吸附的Zn(OH)+中的Zn 原子的作用距离分别为0.202 7、0.192 5 nm,形成了稳定的五元环螯合结构。总的来看,Zn(OH)+预先作用在锡石(110)面后,有利于BHA 在锡石(110)面形成稳定的五元环螯合结构进行吸附。
3 结 论
(1)锡石(110)面的态密度分析结果显示,在费米能级附近主要由O(顶位)原子的2p 轨道组成,说明O(顶位)原子的活性较强,容易参与反应。
(2) 以Zn(OH)+代表的锌组分均可在锡石(110)面以不同形式与O 原子位点间发生吸附作用,Zn 原子与O 原子间均发生电荷转移,轨道向低能量方向移动,形成Zn—O 键;在双桥形式吸附时,Zn 原子的3d 轨道移动更负,转移的电荷量更多,形成的Zn—O 键稳定性更好。说明了Zn(OH)+在锡石(110)面上主要是以面螯合吸附的双桥形式吸附。
(3)BHA 可自发通过羟肟酸基团中的双键O 原子与锡石(110)面的Sn 原子吸附连接成键,然而受空间位阻影响,BHA 分子吸附在锡石表面上未形成稳定的螯合结构。加入锌组分对锡石(110)面作用后,BHA 中双键O 原子和—OH 基团与锡石(110)面上吸附的Zn(OH)+中的Zn 原子形成稳定的五元环螯合吸附,提高了BHA 在锡石(110)面的吸附能力和稳定性。
