单一及复合环境激素对土壤微生物多样性及功能的影响
2023-09-18李仕蓉乔志伟
刘 超, 李仕蓉, 乔志伟
(安顺学院 资源与环境工程学院, 贵州 安顺 561000)
0 引言
【研究意义】环境激素,也被称为环境内分泌干扰物,是一组高度稳定和持久的有机化合物,难以被微生物降解[1]。土壤中的微生物对于土壤生态系统的构建非常重要[2-4],土壤微生物是土壤中生物活性的重要组成部分,可反映土壤环境质量的情况[2]。土壤中的微生物以细菌为主,而细菌群落中含有大量具有一定功能的生理类群,如硝化细菌、氨化细菌、固氮菌等[4-5]。土壤中细菌种类或丰度的变化可直接或间接影响某些生理类群的丰度,从而影响土壤肥力。环境激素进入土壤中,可能对土壤中微生物产生影响,进而影响土壤环境质量导致农作物减产,弄清微生物对环境激素胁迫的响应机制至关重要。【前人研究进展】目前,关于环境激素对微生物的影响研究较多,已有学者研究了不同环境激素下土壤细菌群落的变化[6-7],如郭杨等[8]通过RAPD法研究发现,PAEs(邻苯二甲酸盐类)复合污染提高了土壤基础呼吸,但降低了土壤微生物多样性;谢慧君等[9]利用BIOLOG和RAPD法研究表明,PAEs的浓度越高,对微生物代谢活性的抑制越大,可能是环境激素抑制了微生物的代谢活性,从而降低了微生物的多样性;刘杰等[10]通过平板稀释法研究表明,环境激素皂甙元在高浓度条件下(80 mg/kg)对土壤中的细菌、真菌、放线菌有明显抑制作用;夏庆兵等[11]利用熏蒸提取-容量分析法研究发现,邻苯二甲酸二(2-乙基己)酯(DEHP)对土壤呼吸强度和土壤微生物生物碳量均有影响,并随着DEHP浓度增加呈先升高后降低的趋势;王鑫宏[12]采用氯仿熏蒸-提取方法研究表明,DEHP进入土壤后会影响土壤微生物活性,对微生物碳量表现为降低-升高-降低-稳定的发展趋势;刘畅等[13]利用荧光定量PCR和DGGE相结合的方法,通过控制不同双酚A(BPA)浓度和通气条件研究发现,不同BPA浓度条件下,微生物丰度指数差异显著,BPA浓度对细菌基因丰度的影响也有显著变化。【研究切入点】以上研究均基于单独的环境激素胁迫,而复合环境激素对土壤微生物群落多样性的研究尚少。【拟解决的关键问题】通过选取环境激素中具有典型代表的2种DEHP和BPA物质,应用16sRNA高通量测序技术,探讨DEHP和BPA单一及复合浓度胁迫对土壤细菌群落的变化,深入分析环境激素对土壤细菌群落多样性和结构的影响,为受环境激素污染土壤的改良和利用提供依据。
1 材料与方法
1.1 试验样品采集
试验地位于贵州省安顺市西秀区,该地区属于亚热带湿润高原季风气候,年平均温度13.2~14.0 ℃,年平均降雨量968~1 309 mm,年平均相对湿度80%,是土壤生物多样性研究的合适取样点。试验选取金刺梨种植地土壤,在其中设置面积为3 m×3 m的样方,以五点取样法采集试验区表层0~20 cm处的土壤。
1.2 试验设计
供试土壤中分别添加双酚A(BPA)、邻苯二甲酸二乙酯(DEHP)2种环境激素并设置不同环境激素浓度。依据2018年生态环境部颁发的土壤环境质量标准-农用地土壤污染风险管控标准(试行)规定的环境激素类浓度标准,试验设置BPA、DEHP、BPA+DEHP 3种处理,每个浓度设置2个梯度,每个梯度设4个重复,共28个样品(表1)。
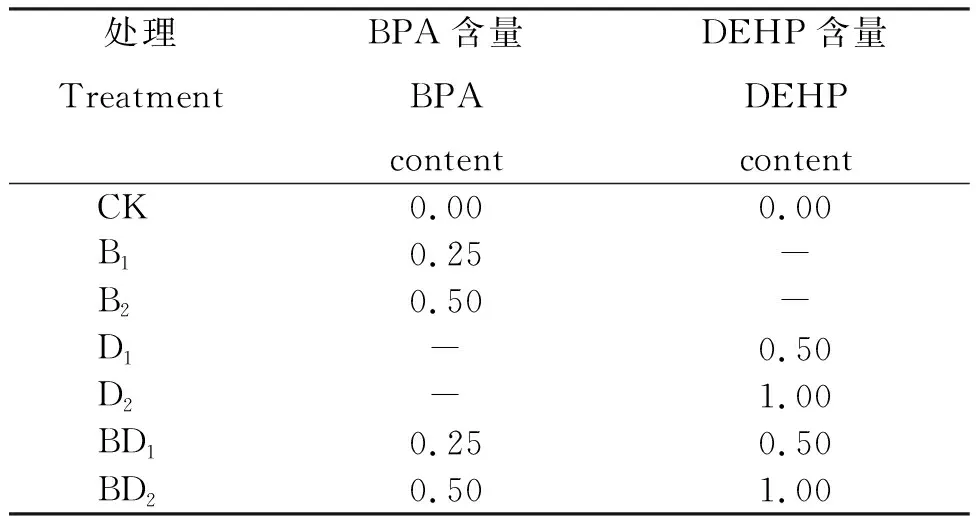
表1 不同处理环境激素设置类型
供试土壤自然风干,碾碎后过2 mm筛。将BPA、DEHP、BPA+DEHP 3种组合浓度溶液加入供试土壤至过饱和状态,对照组加同体积去离子水,每天1次,连续处理7 d。将处理后的土壤风干,碾碎后过2 mm筛,取样并送公司测序。
1.3 DNA抽提及PCR扩增
采用CTAB法提取DNA,用1%琼脂糖凝胶电泳检查提取DNA的纯度和浓度;将适量的DNA放入离心管,用无菌水稀释至1 ng/μL。使用引物341F(5′- CCTAYGGGRBGCASCAG-3′)和806R(5′-GGACTACNNGGGTATCTAAT-3′)对样品的V3和V4区域进行PCR扩增,PCR体系(30 μL):高保真DNA聚合酶Master Mix(2×)15 μL,前后引物各(1 μmol/L)1 μL,gDNA(1 ng/μL)10 μL,ddH2O 3 μL。PCR反应程序:98 ℃预变性1 min;98 ℃变性10 s,50 ℃退火30 s,72 ℃延伸30 s,30个循环;72 ℃延伸5 min。
1.4 Illumina Novaseq 测序
根据获得的PCR产物浓度,将PCR产物进行等量混合,充分混合,利用浓度为2%的1×TAE进行琼脂糖凝胶电泳,然后用Qiagen公司的Qiagen凝胶回收试剂盒对目标条带进行凝胶回收。文库的构建采用Illumina公司TruSeq®DNA PCR-Free Sample Preparation Kit试剂盒,构建的文库经过Qubit定量和文库检测合格后,使用Nova Seq 6000 PE250进行上机测序。
1.5 生物信息学分析
将测序获得的原始fastq序列文件导入QIIME2进行后续处理,然后用QIIME2 dada2插件进行质控、修剪、去噪、拼接及去除嵌合体后得到最终的特征序列(ASV)表格。将获得ASV代表序列导入GREENGENES数据库,根据数据库注释结果得到物种的分类信息表。采用ANOVA和DEseq2法鉴定7个分组和各分组样本间基因丰度有差异的细菌。采用Alpha多样性指数observed OTUs、ACE、Chao1、Shannon、Simpson和Coverage评估样本的多样性程度。Beta多样性指数采用Bray Curtis法估计样本之间的微生物群落结构差异性,并用PCA和NMDS图展示。物种组成多样性热图应用R软件包“ggplot2”绘制,以探究不同处理下微生物群落内物种组成差异情况,最后使用PICRUSt 预测微生物群体可能的功能组成,使用Excel 2019对数据进行前期统计分析,使用SPSS 25.0对数据进行差异显著分析。
2 结果与分析
2.1 细菌的OUT
对28个样品金刺梨种植地土壤中16SrRNA的V3~V4区进行测序共获得2 381 777条原始序列,过滤后获得2 230 088条有效序列,平均每个样品的有效序列数为79 646条,质控后获得56 358条特征序列(ASV),质控效率为70.76%。将ASV的代表序列比对到99%相似度的GREENGENES数据库,共得到46 214个OTUs。注释到门、科、属水平结果分别为98.37%、58.45%和20.18%。通过绘制稀释曲线(图1)看出,随着测序深度的增加28个样品的曲线逐渐趋向于平坦,并在测序量达45 000时获最大值,表明,采集的样品数量足够多,OUTs覆盖率高,测序深度基本覆盖了样品中的所有物种,反映了7个样品的细菌群落和多样性,可以进行后续分析。

图1 OTUs稀释曲线
由图2可见,7组样品中共有OUTs 793个。B1、B2、D1、D2、BD1和BD2分别共有1 409个、1 014个、1 588个、1 621个、1 488个和1 353个OUTs。7组样品的微生物多样性在OUTs存在差异,与CK相比,施用环境激素后土壤中OUTs总数目均减少,B1、B2、D1、D2、BD1和BD2组分别减少43.73%、59.50%、36.58%、35.26%、40.58%和45.97%,表明BPA比DEHP和复合浓度对土壤微生物的影响更大,复合浓度比DEHP对土壤微生物的影响大,总体看,环境激素的施用减少了土壤中微生物种类的数量。

图2 不同样品OUTs花瓣图
2.2 土壤细菌的丰富度及多样性
如表2所示,施用不同浓度环境激素后,样本细菌群落丰富度和多样性均有显著变化。与CK相比,6个处理组的ACE和Chao1指数均显著降低,说明环境激素降低了微生物群落的相对丰度;D2处理组Shannon指数与CK无显著差异,但其他5个处理组的Shannon指数均显著低于CK;D1和D2处理组Simpson指数与CK无显著差异,剩余4个处理组与CK有显著差异。即,BPA组ACE和Chao1指数低于DEHP组和组合浓度组,BD2组ACE和Chao1指数低于B2和D1、D2组,且有显著性差异;BPA组Shannon指数低于DEHP和复合组,复合组Shannon指数低于D2组且有显著性差异。另外7组样品的Coverage指数均接近于最大值1,反映该次测序深度符合试验要求并代表了样本中微生物的真实情况。总体看,由于环境激素对微生物的毒害作用,致使处理组微生物丰富度和多样性降低。

表2 土壤细菌丰富度指数及多样性指数
2.3 土壤微生物群落结构
2.3.1 门水平 由图3可知,不同组别土壤中占据优势地位的菌群主要包括变形菌门(Proteobacteria,占31.37%)、酸杆菌门(Acidobacteria,占29.67%)、放线菌门(Actinobacteria,占10.28%)、绿弯菌门(Chloroflexi,占9.81%)、芽单胞菌门(Gemmatimonadetes,占6.72%),其相对丰度大于5%,约占总序列的87.75%,其次是泉古菌门(Crenarchaeota,占2.42%)、WPS_2(Camdidatus Eremiobacterota,占2.28%)、拟杆菌门(Bacteroidetes,占1.89%)、厚壁菌门(Firmicutes,占1.71%),平均相对丰度均大于1%;其余门类的相对丰度均小于1%,包括疣微菌门(Verrucomicrobia,占0.82%)、消化螺旋菌门(Nitrospirae,占0.67%)等。在不同环境激素浓度条件下,各组微生物的群落组成发生了变化,环境激素胁迫处理显著影响土壤细菌门水平群落结构。各处理组间,B1、B2组中厚壁菌门(Firmicutes)、泉古菌门(Crenarchaeota)、浮霉菌门(Planctomycetes)比D1、D2、BD1、BD2组多;BD1、BD2组中WPS_2比其余处理组显著增多;D2组中放线菌门(Actinobacteria)、消化螺旋菌门(Nitrospirae)比B1、B2、BD2、D1组中显著增多。与CK相比,处理组土壤细菌中的蓝藻门(Cyanobacteria)、拟杆菌门(Bacteroidetes)、疣微菌门(Verrucomicrobia)、变形菌门(Proteobacteria)、芽单胞菌门(Gemmatimonadetes)、迷踪菌门(Elusimicrobia)、装甲菌门(Armatimonadetes)、绿弯菌门(Chloroflexi)减少,而消化螺旋菌门(Nitrospirae)、厚壁菌门(Firmicutes)、酸杆菌门(Acidobacteria)、泉古菌门(Crenarchaeota)增多。说明,在消化螺旋菌门、酸杆菌门、厚壁菌门、绿弯菌门、泉古菌门中可能存在大量对环境激素敏感的群落,在环境激素刺激下大量出现,导致以上3个门占比较CK高,群落结构复杂。
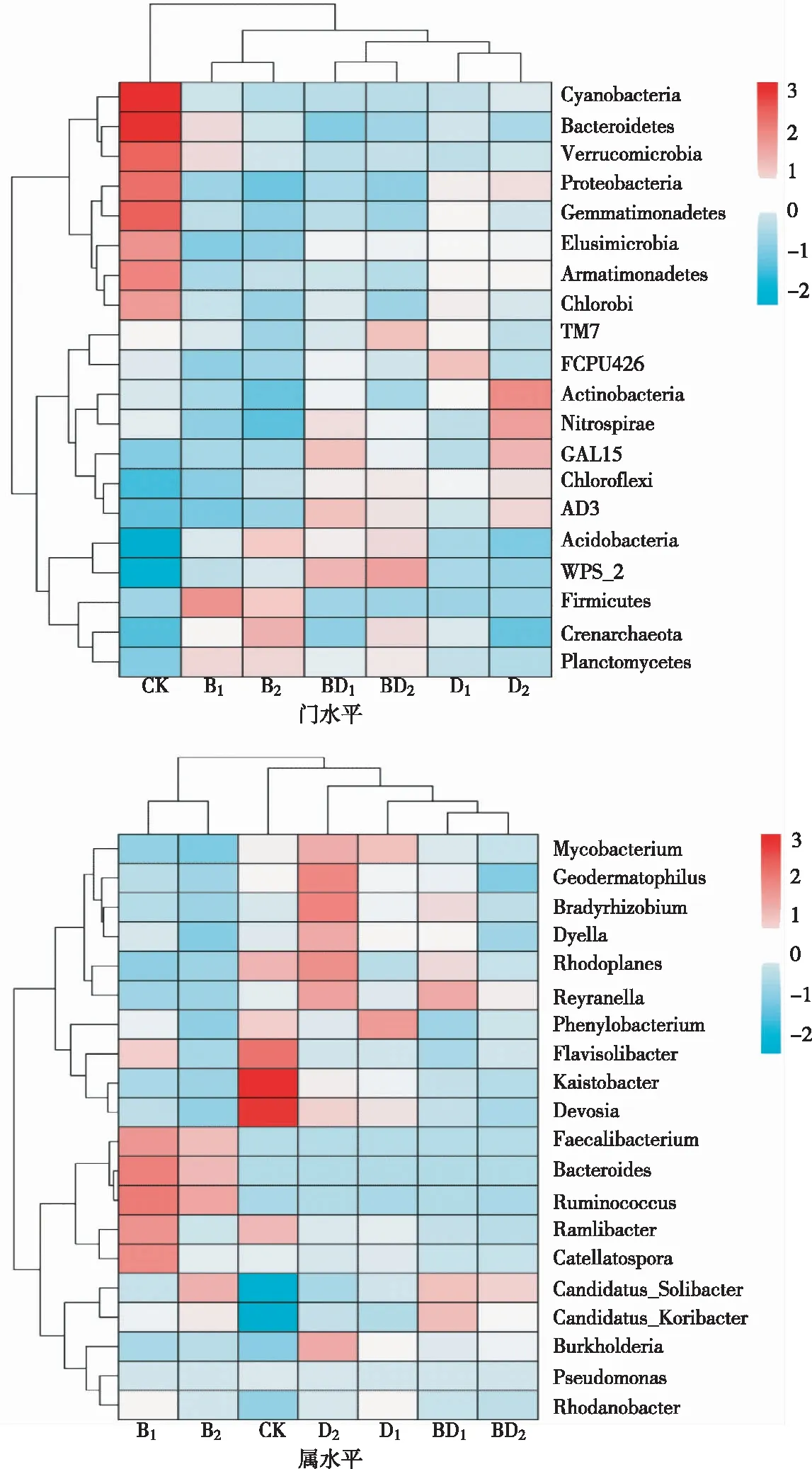
图3 土壤细菌群落聚类分析热图
2.3.2 属水平 由图3可见,在属的水平上,通过序列比对确定每个样品中相对丰度较高的前20个属,其中,念珠菌固体杆菌属(Candidatus_Solibacter,3.09%)、凯氏杆菌属(Kaistobacter,2.64%)、红游动菌属(Rhodoplanes,1.70%)、柯氏假丝酵母菌属(Candidatus_Koribacter,1.68%)4个属的平均相对丰度大于1%,占样品中总序列的9.11%。不同环境激素处理土壤主要细菌属的相对丰度差异显著,与CK相比,施用环境激素的6个土壤处理组中黄色土源菌属(Flavisolibacter)、凯氏杆菌属(Kaistobacter)、德沃氏菌属(Devosia)显著减少,而念珠菌固体杆菌属(Candidatus_Solibacter)、柯氏假丝酵母菌属(Candidatus_Koribacter)、伯克霍尔德菌属(Burkholderia)相对丰度显著高于CK。各处理间种属组成也有差异,B1、B2组中普氏栖粪杆菌属(Faecalibacterium)、拟杆菌属(Bacteroides)、瘤胃球菌数属(Ruminococcus)、链孢菌属(Catellatospora)比D1、D2、BD1、BD2组显著增多,BD1、BD2组中念珠菌固体杆菌属(Candidatus_Solibacter)比其余4个处理组多,D1、D2组中分歧杆菌属(Mycobacterium)、伯克霍尔德菌属(Burkholderia)比B1、B2、BD1、BD2处理组显著增多。
2.4 不同分析方法土壤微生物群落结构
2.4.1 主成分PCA分析法 由图4可见,轴1(PC1)、轴2(PC2)贡献值和总贡献值分别为15.12%、7.47%和22.59%。6个处理组均与CK距离较远,说明经过环境激素处理后试验组与CK群落结构差异较大,同时D1、D2、BD1、BD2各组间距离较远,表明群落结构发生了变化,但B1与B2组间点有部分重合,距离较近,群落相似性高,群落组成比较接近。说明,刺梨种植地土壤经不同环境激素浓度处理后土壤的细菌群落结构有较大改变。
2.4.2 非度量多维尺度分析法 由图5显示,细菌群落结构受不同环境激素显著影响(Stress=0.04),各处理组与CK的距离较远,说明处理组与对照组间微生物群落结构差异度较大;各处理组间,D1与D2组间距离较远,群落相似度低,而B1与B2、BD1与BD2组间距离较近,表明其群落相似度较高,而单一因素BPA组与单一因素DEHP组以及复合浓度组间距离较远,说明单一浓度与复合浓度组间的微生物群落结构差异度较大,群落相似度较低,这与前面的PCA主成分分析结果相同。表明,在不同环境激素浓度处理下,土壤微生物群落结构发生改变,与对照组相比,试验组群落结构改变较大,说明在环境激素的刺激下,土壤中微生物的丰富度和均匀度均发生改变,导致微生物群落结构改变。
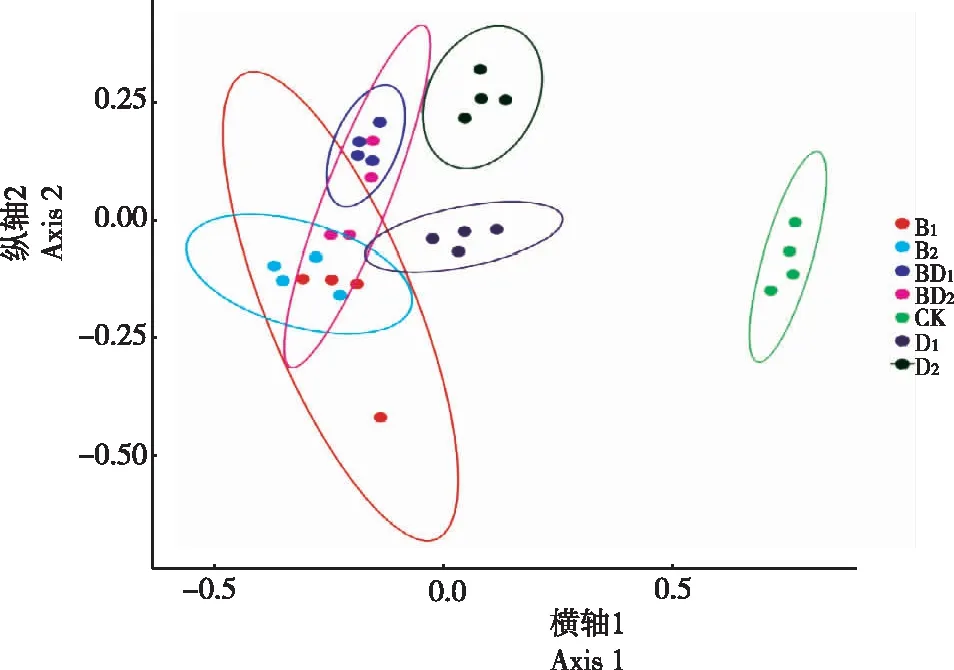
图5 非度量多维尺度分析不同处理土壤微生物的群落结构
2.5 土壤细菌菌群功能
由图6可见,氨基酸代谢(Amino acid metabolism)、辅因子和维生素代谢(Metabolism of cofactors and vitamins)、碳水化合物代谢(Carbohydrate metabolism)、其他氨基酸代谢(Metabolism of other amino acids)、次生产物代谢的生物合成(Biosynthesis of other secondary metabolites)、脂质代谢(Lipid metabolism)、全局概览图(Global and overview maps)、异生物素的生物降解和代谢(Xenobiotics biodegradation and metabolism)等20个子功能预测基因占比在7个样品分组中约为90%,其中,氨基酸代谢、辅因子和维生素代谢、碳水化合物代谢、其他氨基酸代谢、次生产物代谢的生物合成、脂质代谢为预测的优势功能基因,占比分别为11.15%、9.38%、9.31%、6.55%、6.35%、6.15%,可见,以上功能主要属于代谢类型。
由图6可见,在三级功能层级,大部分功能属于未知,约占总和的70.57%,在已知的预测基因功能主要跟生物合成相关,其功能基因丰度在每个样品中均大于1%,丰度总和约为16%,其中萜类化合物和类固醇的生物合成、缬氨酸,亮氨酸和异亮氨酸生物合成、细菌趋药性、脂肪酸合成、酮体的合成和降解为预测的优势功能基因,占比分别约为2.39%、2.14%、1.70%、1.68%和1.65%,其余大部分预测功能基因与物质代谢如生物素代谢、硫辛酸代谢、氨基酸及其衍生物代谢、遗传信息和细胞运动相关。
3 讨论
近年来,由PAEs和BPA引起的环境污染问题已受到广泛关注,我国农田土壤高浓度环境激素检出率增高[13-20]。曹龙等[21]研究发现,珠三角地区的农业土壤中PAEs的平均浓度为21.03 mg/kg;杨彦等[22]发现江苏南部太湖流域农田土壤中15种PAEs总浓度为44.56 mg/kg;夏炎等[23]对广东省6个市的农耕土壤进行调查发现,四溴双酚A平均含量为0.61 ng/g DW,农耕土壤中的四溴双酚A平均蓄积量为4.57 t。环境激素通过不同途径进入土壤中长期残存将严重影响土壤肥力以及农业的可持续发展,可造成农产品病变、失绿、营养元素丢失,甚至导致农作物死亡等。土壤微生物与土壤肥力密切相关,对土壤的形成与发育过程中物质循环和能量转化起至关重要的作用,有利于保持土壤生态系统功能,是土壤质量的重要指标之一[24]。环境激素的施用影响土壤中微生物活动,微生物之间的种间关系、生物量和生物活性会产生一定程度的降低,从而降低土壤肥力。张建等[25]研究表明,PAEs进入土壤中会影响β-葡萄糖苷酶、磷酸酶、脲酶、蛋白酶活性,降低土壤环境质量。研究结果表明,不同浓度环境激素对细菌α-多样性ACE和Chao1指数显著降低,除D2处理组Shannon指数与CK无显著差异外,其余5个处理组Shannon指数均低于CK并存在显著差异,D1和D2组 Simpson指数与CK无显著差异,剩余4个处理组均和CK有显著差异;BPA组ACE和Chao1指数低于DEHP组和组合浓度组,BD2组ACE和Chao1指数低于B2和D1、D2组,且有显著性差异;BPA组Shannon指数低于DEHP和复合组,复合组Shannon指数低于D2组,且有显著性差异。总体看,环境激素对微生物多样性影响最大的是单一BPA,其次是BD复合环境激素,最后是单一DEHP。BPA和DEHP作为增塑剂主要用于塑料包装中,DE SOUZA等[26]研究表明,微塑料中的增塑剂等有害物质进入土壤中,抑制土壤中细菌的生长。高分子量的DEHP在土壤中不容易降解,而相对分子量比较低的BPA可穿过细菌的细胞膜从而被细菌吸收和降解。因此,DEHP处理组比BPA和BD复合组微生物丰度增加。
细菌群落β-多样性分析表明,单一及复合环境激素处理土壤细菌群落结构差异显著。总体上,不同处理土壤细菌优势门类为变形菌门(Proteobacteria,占31.37%)、酸杆菌门(Acidobacteria,占29.67%)、放线菌门(Actinobacteria,占10.28%)、绿弯菌门(Chloroflexi,占9.81%)、芽单胞菌门(Gemmatimonadetes,占6.72%),其相对丰度大于5%,约占总序列的87.75%,这与张慧敏等[27]的研究结果类似;同时PCA、NMDS聚类分析表明,在不同环境激素的刺激下,土壤中微生物的丰富度和均匀度均发生了改变,单一BPA环境激素中厚壁菌门、泉古菌门、浮霉菌门和拟杆菌属丰度较高,单一DEHP中放线菌门、消化螺旋菌门和分歧杆菌属、伯克霍尔德菌属丰度较高,BD复合环境激素中WPS-2和念珠菌固体杆菌属丰度较高,微生物群落结构发生改变,这与解开治等[28]的研究结果相同。不同浓度激素组合处理下细菌门类水平分布特征与已有的研究结果类似[29],刘沙沙等[30]的研究也表明,在微塑料-多环芳烃污染条件下聚氯乙烯-芘改变了菌群的代谢进程,引起了微生物群落结构和功能的改变。这可能是环境激素污染物进入土壤后降低了土壤微生物群落的生理代谢活性和碳源利用能力,从而降低了微生物的多样性、改变了微生物群落结构。
研究结果表明,在二级功能层面,微生物功能以氨基酸代谢、辅因子和维生素代谢、碳水化合物代谢、其他氨基酸代谢、次生产物代谢的生物合成、脂质代谢为预测的优势功能基因;而在三级功能层面已知的预测的基因功能主要跟生物合成相关,跟生物合成相关的功能基因丰度在每个样品中均大于1%,丰度总和约为16%,其余大部分预测功能基因与物质代谢如生物素代谢、硫辛酸代谢,氨基酸及其衍生物代谢、遗传信息和细胞运动相关。靳晓拓等[31]研究表明,对芒果园土壤施用多效矬后,在二级功能层面以碳水化合物代谢、次生产物代谢的生物合成、氨基酸代谢等为主;蒙小俊等[32]对汉江上游污水处理厂的微生物多样性和功能预测研究表明,氨基酸代谢、碳水化合物代谢、次生产物代谢的生物合成以及蛋白质的新陈代谢、遗传信息和物质代谢等有关,这与本研究结果一致。通过16S RNA高通量测序技术方法并运用PICRUSt软件进行微生物功能预测分析,揭示了单一及复合环境激素胁迫下土壤微生物的多样性、结构及其功能组成,但鉴于PICRUSt功能预测分析的局限性和土壤环境的复杂性,后续需要结合土壤环境因子和宏基因组测序技术作进一步分析。
4 结论
单一及复合环境激素进入土壤后显著减少土壤OTUs数量,土壤细菌群落Alpha多样性显著降低,总体看,ACE指数、Chao1指数、Shannon指数和Simpson指数具体表现为BPA组
