蓝细菌CRISPRa系统的开发及其代谢工程应用
2023-09-16王甜甜朱虹杨琛
王甜甜,朱虹,杨琛
(1 中国科学院合成生物学重点实验室,中国科学院分子植物科学卓越创新中心,上海 200032; 2 中国科学院大学,北京 100049)
蓝细菌是光合作用研究的模式生物之一,它们与高等植物一样能够进行产氧光合作用、固定CO2,为生物圈提供重要的初级生产力。蓝细菌的光合固碳过程,可以将太阳能和CO2直接转化为燃料和化学品,同时起到固碳减排和生物合成的作用,因此引起了重点关注。通过对蓝细菌进行代谢工程改造,已经实现从CO2到数十种燃料及化学品的生物合成,展示出蓝细菌光合生物技术的重要潜力[1‑4]。
近年来蓝细菌遗传操作工具的开发取得了重要进展,包括基因表达调控相关的启动子与核糖体结合位点的挖掘以及核糖开关的应用[5‑9]。然而与大肠杆菌(Escherichia coli)等模式微生物相比,现有蓝细菌的遗传操作工具仍然较为缺乏且效率较低,因此开发高效的蓝细菌基因调控工具对于蓝细菌系统与合成生物学研究具有重要意义。随着CRISPR 技术的发展,它在蓝细菌中的应用也受到了关注。目前在蓝细菌中已建立了基于具有DNA 双链切割活性的Cas9 或Cas12a 的CRISPR 基因编辑技术,实现基因的插入、删除及碱基替换[10‑12]。研究者们也利用失去DNA 切割功能的dCas9(dead Cas9)或dCas12a(dead Cas12a)在蓝细菌中建立了抑制基因表达的CRISPR 干扰(CRISPR interference,CRISPRi)系统[13‑14],即引导dCas9 或dCas12a 至目标基因的转录起始位点(transcription start site,TSS),则dCas9 或dCas12a会物理性阻碍RNA聚合酶的通过,导致基因沉默。Yao 等[13]在集胞藻Synechocystissp. PCC 6803 中建立了基因组规模的CRISPRi文库,并用于乳酸耐受和高产相关基因的挖掘。然而,蓝细菌中激活基因表达的CRISPR(CRISPR activation, CRISPRa)系统尚未建立。事实上,细菌中CRISPRa 技术的报道仍然较少,目前只有在大肠杆菌、枯草芽孢杆菌(Bacillus subtilis)、产酸克雷伯氏菌(Klebsiella oxytoca)等少数几种模式细菌中建立了该技术,主要是将dCas9和转录激活因子结合,靶向目标基因启动子上游序列,通过招募或稳定RNA聚合酶,从而激活目标基因的转录[15‑17]。对于CRISPRa系统中的转录激活因子,目前已报道的包括RNA聚合酶ω‑亚基RpoZ[15‑16,18‑20]、氧化应激转录调控蛋白SoxS(superoxide response transcriptional regulator)[21‑23]、抗Sigma 因子AsiA[24]以及σ54因子依赖型激活蛋白[17]。本研究在模式蓝细菌聚球藻(Synechococcus elongatus)PCC 7942(以下简称为PCC 7942)中筛选转录激活因子,建立了CRISPRa系统,并从靶向位点等多个方面进行测试对该系统进行了优化。
异戊烯醇(isopentenol)包括3‑甲基‑3‑丁烯‑1‑醇(isoprenol)和3‑甲基‑2‑丁烯‑1‑醇(prenol),具有高研究法辛烷值(reaserch octane number,RON)和良好的燃烧特性,可作为汽油替代品,是一种重要的生物燃料。Wither 等[25‑30]在大肠杆菌(Escherichia coli)中实现了异戊烯醇的异源合成。本研究以异戊烯醇在PCC 7942 中的异源合成为研究对象,测试了构建的CRISPRa 系统能否用于蓝细菌代谢工程,结果展示了该系统能够用于多基因代谢途径的优化改造,大幅提高了PCC 7942 中异戊烯醇的生物合成。
1 材料和方法
1.1 试剂和仪器
1.1.1 试剂
3‑甲基‑3‑丁烯‑1‑醇、3‑甲基‑2‑丁烯‑1‑醇、硫酸铁铵、3‑甲基‑2‑苯并噻唑啉酮腙盐酸盐水合物、氨基磺酸、溶菌酶,均购自西格玛奥德里奇(上海)贸易有限公司;RNA 抽提试剂盒,购自德国Macherey‑Nagel 公司;RNA 反转录试剂盒,购自TOYOBO;多片段一步法快速克隆试剂盒、DEPC水、qPCR SYBR Green Master mix,均购自翌圣生物科技(上海)股份有限公司;质粒抽提试剂盒、胶回收试剂盒、细菌基因组抽提试剂盒,均购自美国AXYGEN 公司;高保真DNA 聚合酶(2×Phanta Max Master Mix),购自南京诺唯赞生物科技股份有限公司;限制性内切酶,购自纽英伦生物技术(北京)有限公司(New England Biolabs);DH5α 感受态细胞,购自深圳康体生命科技有限公司。
1.1.2 仪器
Biometra TAdvanced Twin PCR 仪,购自耶拿分析仪器(北京)有限公司;Eppendorf Centrifuge 5417R 冷冻离心机,Eppendorf Centrifuge 5418、5430 高速离心机,均购自德国艾本德股份公司;BioTek power wave XS2 全波长酶标仪,购自贝克曼库尔特国际贸易(上海)有限公司;TECAN Spark 多功能微孔板检测仪,购自帝肯Tecan 上海贸易有限公司;Agilent 7890A 气相色谱仪,购自安捷伦科技(中国)有限公司;Bio‑Rad MYIQ2荧光定量PCR 仪,购自伯乐生命医学产品(上海)有限公司;Thermo Scientific NANODROP2000C 微量分光光度计,购自赛默飞世尔科技(中国)有限公司;复日科技FR‑980A 凝胶成像仪,购自上海复日科技有限公司;GXZ‑280C 智能光照培养箱,购自宁波江南仪器厂。
1.2 基因克隆和质粒的构建
提取集胞藻PCC 6803 基因组,设计引物(P18082‑F 与P18082‑R,P18082upstream‑F 与P18082upstream‑R)进行PCR 得到启动子P18082及其上游序列,通过同源重组的方法使用多片段一步法快速克隆试剂盒将其与绿色荧光蛋白基因sfgfp以及质粒载体PCL1920‑NSⅡ(以PLC1920 质粒为骨架[1],在SalⅠ与SacⅠ酶切位点处引入PCC 7942 中NSⅡ中性位点的同源臂序列)进行连接,得到质粒P01,然后将不同的sgRNA基因序列及其启动子Pcpc(sgRNA 及启动子Pcpc 由南京金斯瑞公司合成)也通过同源重组的方法引入该质粒,得到含有不同靶位点sgRNA的质粒(P02‑P07)。
提取大肠杆菌E.coli基因组,以此为模板,设计引物(nudⅠ‑F 与nudⅠ‑R)进行PCR 得到nudⅠ(GenBank:UZX07358.1);使用限制性内切酶EcoRⅠ和BsrGⅠ酶切质粒P05,通过同源重组将nudⅠ连接到酶切后的质粒载体上,得到质粒P08;提取PCC 7942 基因组,以此为模板,设计引物(dxs‑F 与dxs‑R)通过PCR 得到dxs片段,与nudⅠ连接后将其分别插入EcoRⅠ和BsrGⅠ酶切后的质粒P05和P07,构建得到质粒P09和P10。
以质粒P09 为基础,将由南京金斯瑞合成的crRNA 和tracrRNA 序列通过同源重组替换P09 质粒中的sgRNA,构建得到质粒P11~P13。
提取化脓性链球菌(Streptococcus pyogenes)基因组,以此为模板,通过设计引物(dCas9‑F 与dCas9‑R)引入两个突变位点(D10A和H840A)进行PCR得到dCas9,设计引物(Pendo‑F与Pendo‑R)通过PCR 得到启动子Pendo;soxS(GenBank:NP_31307)、RNA 聚合酶ω‑亚基rpoZ(GenBank:ABB57740.1)、α‑亚基rpoA(UniProt/Swiss‑Prot:Q31L30.1)、RpoA 的N 端结构域(UniProt/Swiss‑Prot:Q31L30.1 中第1~681 位碱基)、Sigma‑70 因子rpoD(GenBank:D10973.1)、dxs(GenBank:CAD55646.1)均由南京金斯瑞公司合成。通过同源重组的方法分别与质粒载体PCL1920‑NSⅢ(以PLC1920质粒为骨架,在SalI与SacI酶切位点处引入PCC 7942 中NSⅢ中性位点的同源臂序列)或PCL1920‑RpoZ(以PLC1920 质粒为骨架,在SalⅠ与SacⅠ酶切位点处引入PCC 7942中rpoZ的同源臂序列)进行连接,构建成含有不同转录激活蛋白的质粒(P14‑P25)。
dCas9 相关质粒通过同源重组双交换整合到PCC 7942 基因组上的NSⅢ和RpoZ 位点,抗性筛选标记为卡那霉素(kanamycin, KmR);sfgfp、nudⅠ、dxs相关质粒通过同源重组双交换整合到PCC 7942 基因组上的NSⅡ位点,抗性筛选标记为壮观霉素(spectinomycin, SpeR)。
1.3 菌株转化和培养方法
(1)蓝细菌的转化方法 取OD730≈1~2 的菌液1 mL,5000 r/min 离心3 min,去上清;加入500 µL的NaCl溶液(10 mmol/L)重悬后5000 r/min离心3 min,去上清;加入10 µL 的BG11[31]培养基重悬菌体,再加入1 µg 质粒,用移液器混匀后用锡箔纸包裹离心管,置于摇床,190 r/min 30 ℃过夜培养后涂于相应抗性平板。培养5~7 d 后,挑取单克隆在相应抗性的BG11 固体培养基上进行传代来纯合基因组,使用验证引物进行PCR 来确认目标基因是否整合到基因组上。
(2)蓝细菌摇瓶培养方法 将蓝细菌接种到含有25 mL BG‑11 培养基(含100 mmol/L NaHCO3)的三角瓶中,三角瓶用纱布封口,光照强度3000 lx,温度30 ℃,摇床转速190 r/min。对于异戊烯醇合成菌株的培养,纱布换成密封性更好的封口膜。
(3)BG‑11培养基(含100 mmol/L NaHCO3)配制 1.5 g/L NaNO3,0.075 g/L MgSO4·7H2O,0.036 g/L CaCl2·2H2O,4.76 g/L HEPES,1 mL/L 溶液1,1 mL/L 溶液2,1 mL/L 溶液3;加入8.4 g/L 的NaHCO3,用NaOH调节pH至7.5,过滤除菌。
溶液1:6.567 g/L 一水柠檬酸C6H8O7·H2O,6 g/L 柠檬酸铁铵C6H8FeNO7,1.107 g/L Na2EDTA·2H2O,过滤除菌。
溶液2:2.86 g/L H3BO3,1.545 g/L MnSO4·H2O,0.222 g/L ZnSO4·7H2O,0.391 g/L Na2MoO4·2H2O,0.0404 g/L CoCl2·6H2O,过滤除菌。
溶液3:52 g/L K2HPO4·3H2O,20 g/L Na2CO3,过滤除菌。
本研究工作所用到的蓝细菌菌株、质粒和引物见表1~表3。

表1 本实验所用到的菌株Table 1 Strains used in this study

表2 本实验所用到的质粒Table 2 Plasmids used in this study

表3 本实验所用到的引物Table 3 Primers used in this study
1.4 RNA提取和实时定量PCR
将蓝细菌培养至OD730为0.7~0.8,取5 mL 悬液,离心收集菌体,加入10 mg溶菌酶进行细胞裂解。使用RNA 提取试剂盒(Macherey‑Nagel)提取总RNA,用反转录试剂盒(TOYOBO)除去DNA 污染并进行RNA 的反转录,得到的cDNA 用实时定量PCR仪(Bio‑Rad)进行定量分析。
1.5 绿色荧光蛋白的检测
菌株的初始OD730为0.1,培养0 h、24 h、48 h、72 h 和96 h 时分别取菌液100 μL,测定OD730以及菌体的荧光值。利用多功能微孔板检测仪(TECAN Spark)进行荧光强度检测,激发波长为485 nm,发射波长为520 nm[32]。以OD730为横坐标、荧光值为纵坐标作标准曲线,得到OD730为1.0时的荧光值,扣除野生型菌株的荧光值后即为该菌株的荧光值。
1.6 显色法检测异戊烯醇
在酶标板中依次加入20 µL 蓝细菌培养液上清或3‑甲基‑3‑丁烯‑1‑醇与3‑甲基‑2‑丁烯‑1‑醇的标准品、50 µL 硫酸铁铵与氨基磺酸的混合液、10 µL 3‑甲基‑2‑苯并噻唑啉酮腙盐酸盐水合物(3‑methyl‑2 benzothiazolinone hydrazine hydrochloride hydrate,MBTH)及120 µL 水,室温反应40 min 后测定620 nm 处的吸光度A620[33]。该方法的标准曲线为:y= 0.091 52x+ 0.014 06;y为A620值,x为异戊烯醇浓度(0~6 mg/L)。
1.7 气相色谱法检测异戊烯醇
用3‑甲基‑3‑丁烯‑1‑醇和3‑甲基‑2‑丁烯‑1‑醇的标准品分别配出不同浓度的标准品。菌株培养液离心后取上清。标准品和样品经气相色谱(gas chromatography;Agilent 7890A)连接火焰离子化检测器(Agilent)进行检测和定量。使用的气相色谱柱(Alltech EC‑WAX)规格为30 m×0.32 mm。进样量1 μL,柱温85 ℃ 3.5 min,150 ℃ 1.325 min,200 ℃ 6 min。
2 结 果
2.1 CRISPRa系统的构建
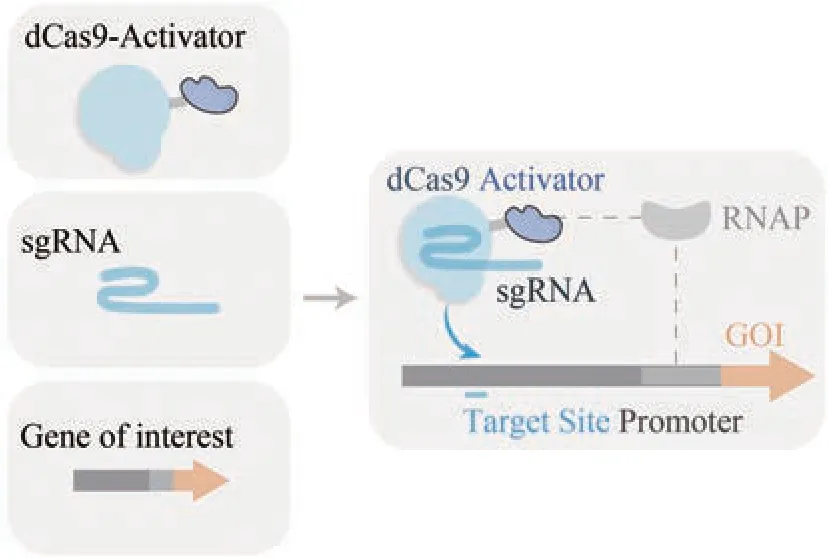
CRISPR 激活系统由dCas9、转录激活因子以及具有向导作用的sgRNA(single guide RNA)组成[图1(a)],形成的复合体招募或者稳定RNA聚合酶于目标基因的启动子区域。将来源于化脓性链球菌(Streptococcus pyogenes)且经过突变得到的dCas9与转录激活因子融合表达,两者之间通过柔性连接肽(CAGGGGSGGGGS)连接,该基因模块插入至PCC 7942 染色体的中性位点NSⅢ上,由化脓性链球菌CRISPR/Cas9 系统的天然启动子Pendo 驱动表达[图1(b)]。sgRNA 和绿色荧光蛋白(sfGFP)基因插入至中性位点NSⅡ上,分别由集胞藻PCC 6803 藻蓝素合成基因(sll1577)的启动子Pcpc 和集胞藻PCC 6803 50S 核糖体蛋白L10(Sll1745)基因的启动子P18082驱动表达。
对于CRISPRa 系统中的转录激活因子,我们分别测试了大肠杆菌来源的转录激活蛋白SoxS、PCC 7942来源的RNA聚合酶ω‑亚基RpoZ、α‑亚基RpoA、RpoA 的N 端结构域(N‑terminal domain,NTD)以及Sigma‑70 因子RpoD,获得其与 dCas9融合表达的菌株G09~G13。荧光检测结果显示这些转录激活因子都能对目标基因的表达起到一定的激活作用,其中表达RpoZ 的菌株G09 的荧光信号值最高,是对照菌株G03(无dCas9 与转录激活因子模块)的3.16倍[图1(c)]。
为了证实CRISPRa 系统的转录激活作用,以外源sgRNA 片段AGGACGCCTTTGGTAACCGC作为脱靶(off‑target)RNA 构建菌株G02 以及缺失sgRNA 的菌株G01。结果显示G09 的荧光信号值显著高于G01和G02菌株,而后两者的荧光信号值没有明显差别[图1(d)]。
2.2 CRISPRa系统的优化
我们测试了CRISPRa 系统在目标基因上游不同的靶向位点[图2(a)],即非模板链上转录起始位点TSS 上游216 bp、236 bp、273 bp 的H1、H2和H3位点以及模板链上TSS上游327 bp、397bp的H4 和H5 位点[16](表4)。结果显示对于TSS 上游273~397 bp 范围内的靶点H3~H5,CRISPRa 系统都具有一定的激活作用,其中H4 是最佳的靶点;靶点H5距离TSS较远,与靶点H4相比转录激活效果降低;靶点H1 距离TSS 最近,目标基因的表达受到抑制[图2(b)]。这些结果表明该系统可以通过靶向启动子上游不同位点,达到激活或者抑制目标基因表达的效果。
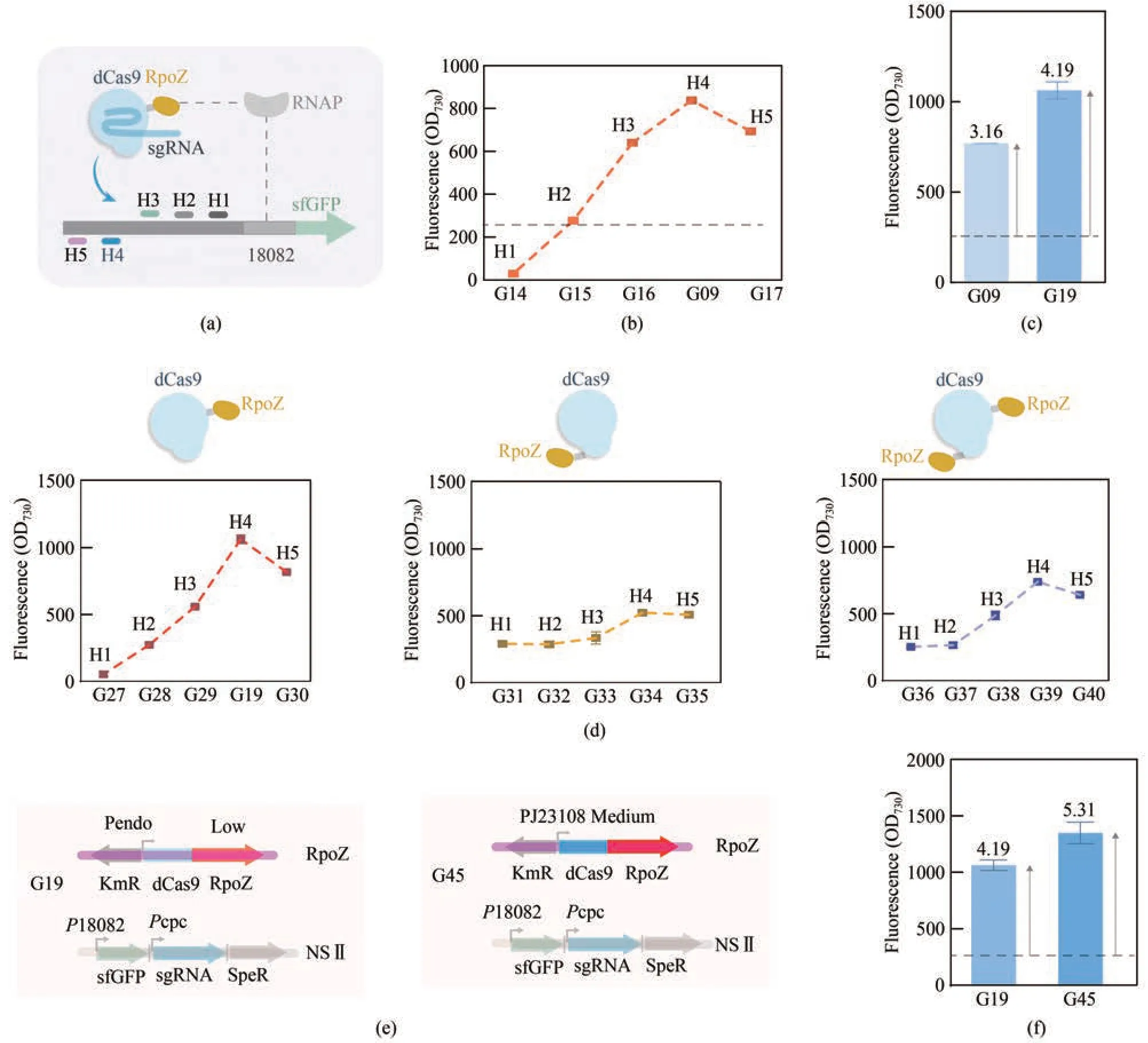
图2 CRISPRa系统的优化(a)目标基因上游不同靶向位点的位置示意图;(b)CRISPRa靶向不同位点的菌株的荧光信号值;(c)内源rpoZ被敲除的G19菌株的荧光信号值;(d)dCas9 与RpoZ 融合表达顺序的改变对于CRISPRa 激活效果的影响;(e)以强启动子PJ23108 替换弱启动子Pendo,构建dCas9‑RpoZ表达增强的菌株G45;(f)菌株G45的荧光信号值Fig. 2 Optimization of the CRISPRa system(a)Scheme of different targeting sites for CRISPRa. (b) The fluorescence intensities of strains G09, G14, G15,G16, and G17 with different targeting sites. (c) The fluorescence intensity of G19 strain with genetic knockout of endogenous rpoZ. (d) Effect of different orientations of the dCas9 and RpoZ fusion on CRISPRa performance. (e) Expression of dCas9‑RpoZ was increased in G45 strain by replacing the promoter Pendo with PJ23108.(f) The fluorescence intensity of G45 strain.
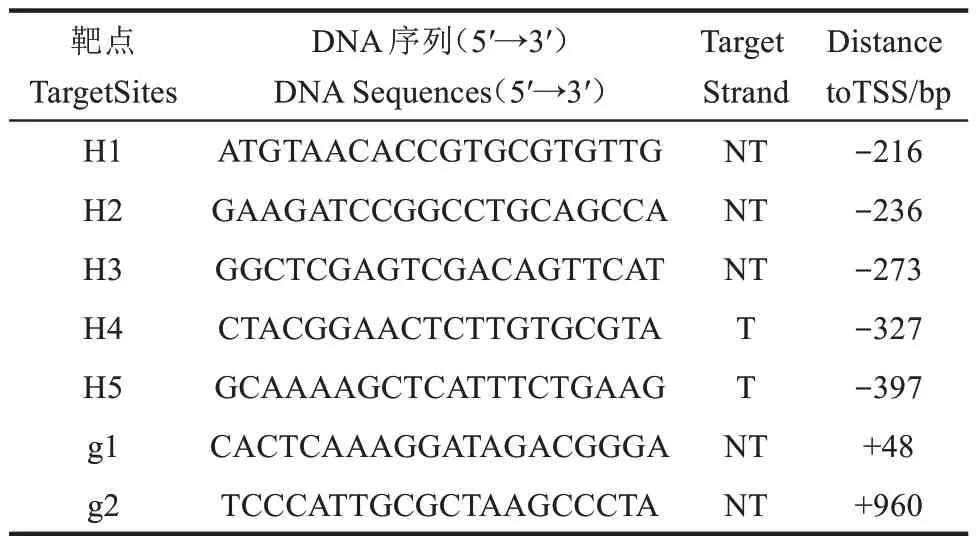
表4 本研究用到的CRISPRa系统靶位点序列Table 4 Targeting sequences for the CRISPRa system used in this study
为了提高CRISPRa 系统的转录激活效果,我们以dCas9‑RpoZ 基因模块替换PCC 7942 基因组上的rpoZ基因,即敲除内源rpoZ基因,得到菌株G19,其荧光信号值显著高于G09 菌株,达到对照菌株G03 的4.19 倍[图2(c)]。因此,敲除内源rpoZ基因有效提高了CRISPRa系统的激活效果。
我们测试了dCas9 与RpoZ 融合表达顺序的改变对于CRISPRa 激活效果的影响。除了RpoZ 融合在dCas9 的C 端(dCas9‑RpoZ),我们还构建了RpoZ 融合在dCas9 的N 端(RpoZ‑dCas9)以及N端和C 端(RpoZ‑dCas9‑RpoZ)。对于每一种构建方式,我们测试了不同的靶向位点。荧光检测结果表明这三种融合表达顺序均在靶点H4 达到最优的激活效果,其中激活效果最佳的构建方式是dCas9‑RpoZ[图2(d)]。
我们还测试了增强dCas9‑RpoZ 的表达对于CRISPRa 激活效果的影响。我们将低强度的启动子Pendo 替换为较高强度的启动子PJ23108,驱动dCas9‑RpoZ 基因模块的表达,得到菌株G45[图2(e)]。该菌株的荧光信号值显著高于G19 菌株,达到对照菌株G03的5.31倍[图2(f)]。
2.3 CRISPRa 系统应用于异戊烯醇合成途径的优化
我们将构建的CRISPRa 系统应用于蓝细菌代谢工程,选择的对象是异戊烯醇(isopentenol)在PCC 7942中的异源合成。异戊烯醇包括3‑甲基‑3‑丁烯‑1‑醇(isoprenol)和3‑甲基‑2‑丁烯‑1‑醇(prenol),这两者可以在Nudix水解酶家族的作用下由甲基赤藓糖醇‑4‑磷酸途径(methylerythritol phosphate,MEP 途径) 上的异戊烯焦磷酸(isopentenyl pyrophosphate,IPP) 和二甲基烯丙基焦磷酸(dimethylallyl diphosphate,DMAPP)分别生成[30]。PCC 7942 中有MEP 途径,但是不能合成异戊烯醇。
2.3.1 异戊烯醇合成基因的表达
我们在PCC 7942 中引入大肠杆菌来源编码核苷三磷酸酶(nucleoside triphosphatase)的nudⅠ基因。CRISPRa 靶向位点为nudⅠ上游H4 位点。sgRNA和nudⅠ插入至PCC 7942染色体的中性位点NSⅡ上,分别由Pcpc 和P18082 驱动表达。dCas9‑RpoZ基因模块替换基因组上的rpoZ基因或者插入至中性位点NSⅢ,分别得到菌株S03和S04[图3(a)]。将菌株在含有NaHCO3的无机盐培养基中光照培养96 h,利用qRT‑PCR 检测菌株中nudⅠ的转录水平,利用显色反应检测培养液上清中异戊烯醇的含量(A620)。结果显示与对照菌株S01(无dCas9‑RpoZ模块)相比,菌株S03和S04中nudⅠ表达水平分别提高12.7 倍和13.4 倍,异戊烯醇的生成明显增加[图3(b)、(c)]。
2.3.2 激活合成途径上两个基因
由dxs基因编码的脱氧木酮糖‑5‑磷酸合成酶(1‑deoxy‑D‑xylulose 5‑phosphate synthase)是MEP途径的限速步骤[图4(a)]。我们利用CRISPRa 对nudⅠ和dxs进行同时激活,得到菌株S05 和S06,其中S06 缺失rpoZ基因,另外用脱靶RNA 构建了菌株S07[图4(b)]。我们检测了这些菌株中nudⅠ和dxs的转录水平以及异戊烯醇的生成。结果显示与对照菌株S02(无dCas9‑RpoZ模块)相比,菌株S05和S06的异戊烯醇产量显著提高,其中S06的产量更高,而菌株S07的产量无明显变化[图4(c)]。菌株S05 和S06 的nudⅠ和dxs的表达被明显激活。与S02 相比,菌株S05 中nudI和dxs的转录水平分别提升了5.96 倍和2.14 倍,菌株S06 中nudⅠ和dxs的转录水平分别提升了7.66 倍和2.80 倍;而菌株S07 中基因表达水平没有明显改变[图4(d)]。因此,该CRISPRa 系统不仅能够高效激活单个基因的表达,而且能同时激活两个基因的表达。

图4 CRISPRa同时激活nuⅠd与dxs(a)dxs 编码的脱氧木酮糖‑5‑磷酸合成酶催化MEP 途径的第一步反应;(b)sgRNA(靶位点H4)、nudⅠ及dxs 整合至PCC 7942 中性位点NSⅡ,dCas9‑RpoZ基因模块整合至PCC 7942中性位点NSⅢ或替换内源rpoZ基因,分别得到菌株S05和S06,S02为没有dCas9‑RpoZ模块的对照菌株,S07为整合了脱靶RNA(off target)的菌株;(c)菌株S02、S05、S06、S07培养96 h后利用显色反应检测的上清液中异戊烯醇含量;(d)菌株S02、S05、S06、S07培养72 h后nudⅠ与dxs的转录水平Fig. 4 Simultaneous activation of nudⅠ and dxs by CRISPRa(a) 1‑Deoxy‑D‑xylulose 5‑phosphate synthase encoded by dxs catalyzes the first reaction of the MEP pathway. (b)Scheme of nudⅠ and dxs activation by CRISPRa. The sgRNA H4, nudⅠ, and dxs were inserted into the NSⅡ site. The DNA fragment encoding dCas9‑RpoZ was inserted into the NSⅢsite or to replace endogenous rpoZ, generating strains S05 and S06, respectively. S02 (without dCas9‑RpoZ) and S07 (with an off target sgRNA 106)are the control strains. (c) Isopentenol production by strains S02, S05, S06, and S07.Cells were cultivated for 96 h, and isopentenol in the culture supernatant was detected by a colorimetric assay. (f) Transcriptional levels of nudI and dxs in strains S02, S05, S06, and S07. RNA was isolated from cells cultivated for 72 h.
2.3.3 激活合成途径基因及抑制竞争途径基因
异戊烯醇合成的前体代谢物IPP 和DMAPP 在香叶基焦磷酸合成酶(geranylgeranyl diphosphate synthase,GPPS)的作用下生成GPP,进而合成类胡萝卜素和叶绿素等复杂萜类化合物[34],因此GPPS 是影响异戊烯醇合成的竞争途径上的关键酶[图5(a)]。我们利用建立的CRISPR 系统对nudⅠ和dxs进行激活,同时也对gpps基因的转录进行抑制。在PCC7942 的基因组上gpps(Synpcc7942_0776)位于一个操纵子的中间[图5(b)]。我们在该操纵子的TSS 以及gpps基因的起始密码子附近各选了一个靶向位点(g1 与g2,见表2),nudⅠ和dxs的上游靶向位点为H4 位点,这样得到菌株S12和S13,另外用脱靶RNA 构建了菌株S14,S12~S14都缺失rpoZ基因[图5(c)]。
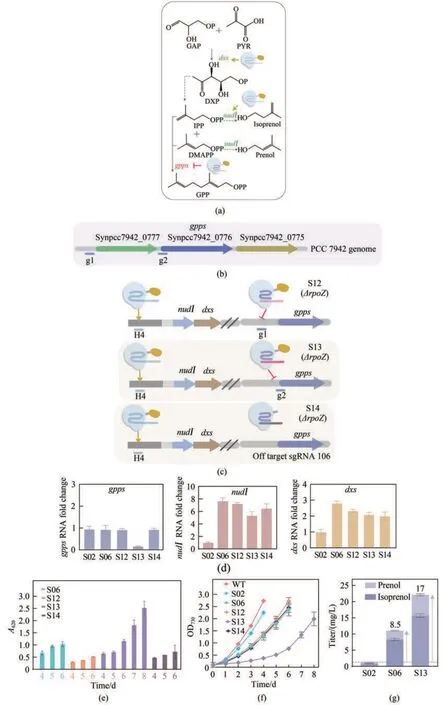
图5 CRISPRa 同时激活nudI与dxs并抑制gpps(a)gpps编码的香叶基焦磷酸合成酶是影响异戊烯醇合成的竞争途径上的关键酶;(b)gpps(Synpcc7942_0776)基因在PCC 7942 基因组上的位置示意图;(c)在S06菌株gpps所在操纵子的转录起始位点以及gpps基因起始密码子附近各选了一个靶向位点(g1与g2),分别得到菌株S12 和S13,S14 为整合了脱靶RNA 的对照菌株;(d)菌株S12、S13、S14 及S02、S06 在OD730约为0.7 时gpps、nudⅠ和dxs的转录水平;(e)菌株S06、S12、S13与S14培养4~8天时利用显色反应检测的上清液中异戊烯醇含量;(f)野生型菌株WT与S02、S06、S12、S13、S14菌株的生长曲线;(g)菌株S02、S06与S13在OD730约为2.0时利用气相色谱检测的培养上清液中异戊烯醇的含量Fig. 5 Simultaneous activation of nudⅠ and dxs and repression of gpps by CRISPRa(a) gpps catalyzes a key competing reaction for isopentenol biosynthesis; (b) Location of the gpps gene and the targeting sites g1 and g2 on the PCC 7942 genome; (c) The g1 and g2 sites of gpps in strain S06 were targeted, generating strains S12 and S13, respectively. S14 (with an off target sgRNA 106) is a control strain. (d) Transcriptional levels of gpps, nudⅠ and dxs in strains S02, S06, S12, S13, and S14. RNA was isolated from cells grown to OD730 of about 0.7. (e) Isopentenol production by strains S02, S06, S13, and S14. Isopentenol in the culture supernatant was detected by a colorimetric assay. (f) Isopentenol production by strains S02, S06, and S13. Culture supernatants were collected at OD730 of about 2.0, and isopentenol was detected by gas chromatography.
我们检测了上述菌株中nudⅠ、dxs及gpps的转录水平以及菌株的生长与异戊烯醇的生成。结果显示与菌株S06相比,菌株S13中gpps的转录水平降低了85%,而菌株S12 和S14 中gpps的表达水平没有明显改变;菌株S13 中nudI与dxs的转录水平与菌株S06 相比降低了25%~30%,但是与对照菌株S02相比仍然提高了5.33倍和2.09倍[图5(d)]。菌株S13 的异戊烯醇产量明显高于菌株S06,在光照培养第8 天达到最高值[图5(e)]。菌株S13 的生长明显变慢[图5(f)],可能是由于gpps基因受到强烈抑制而导致光合作用相关的叶绿素和类胡萝卜素合成下降。除了显色法以外,我们还利用气相色谱检测了菌株的异戊烯醇生成。结果显示菌株S13 的异戊烯醇产量达到22.1 mg/L,相比菌株S06 和S02 分别提高了2 倍和17 倍[图5(g)]。因此,该CRISPRa 系统能够激活nudⅠ与dxs的转录并同时抑制gpps基因的转录,大幅提高了蓝细菌中异戊烯醇的合成。
3 总结与讨论
本研究以光合作用模式生物、光能自养细胞工厂底盘--蓝细菌为研究对象,开发了CRISPR激活系统。利用高强度启动子表达sgRNA,将dCas9 与多个不同的转录激活因子融合表达,发现RpoZ 是较好的转录激活因子。进而通过内源rpoZ基因敲除以及dCas9‑RpoZ 靶向位点优化与表达增强,对该CRISPRa 系统进行了优化。利用建立的CRISPRa 系统对重要生物燃料--异戊烯醇的合成途径进行了工程改造,该系统能够实现单基因的高表达、多基因的同时激活以及不同基因的同时激活和抑制,大幅提高了蓝细菌中异戊烯醇的合成,展示了该系统能够有效地用于多基因代谢途径的优化改造,有望成为光能自养细胞工厂构建的有力工具。
除了代谢工程应用以外,该CRISPRa系统的另一个潜在应用是通过建立gRNA文库,构建蓝细菌基因组规模的基因表达扰动文库,进行功能获得性的遗传筛选(gain‑of‑function screening),用于挖掘能够赋予特定细胞表型的基因,比如促进生长、增强耐受性和化学品生产相关的基因等。在此之前,需要测试该CRISPRa系统在基因组原位调控目标基因的表达。建立CRISPRa文库,高通量地进行功能基因组学研究和功能元件挖掘,这是CRISPRa技术与非CRISPR 的基因调控技术相比较的一大优势。哺乳动物细胞中的CRISPRa文库已有报道[35‑41],被用于功能获得型筛选,如耐药机制研究。然而,细菌中的CRISPRa文库尚未被报道。
目前只有在大肠杆菌等少数几种模式细菌中建立了CRISPRa技术,本研究可以为其他非模式菌株中CRISPRa系统的开发提供思路。对于系统中的转录激活因子,本研究主要测试了蓝细菌的RNA聚合酶亚基等。为了提高该CRISPRa 系统的激活效果,需要测试更多的蓝细菌内源或外源的转录激活因子,dCas9 以外的其他CRISPR 蛋白(比如dCpf1),还需要测试目标基因上游更多的靶向位点,结合生物信息学分析等实现最优靶点的预测。
