盐酸厄洛替尼的合成
2023-09-11陈宁
陈 宁
(海南建科药业有限公司,海南 海口 571200)
厄洛替尼,化学名:N-(3-乙炔苯基)-6,7-双(2-甲氧乙氧)-4-喹啉胺,商品名为特罗凯,临床用其盐酸盐。通过临床试验,厄洛替尼于2004年1月由美国FDA批准上市,2005年9月,厄洛替尼率先在欧盟获准用于胰腺癌和非小细胞肺癌二、三线的治疗,2007年在中国上市,用于晚期局限性、转移性非小细胞肺癌病人的二线或三线治疗[1-3]。除了治疗非小细胞肺癌外,还与吉西他滨联合用于治疗晚期胰腺癌。
EGFR酪氨酸激酶抑制剂耐受性好,不良反应少,通过抑制受体活性有效抑制肿瘤的发生和发展,因此以酪氨酸激酶为靶点的抗肿瘤治疗成为癌症中十分活跃的研究领域,对酪氨酸激酶的小分子抑制剂的研究有了很大的进展[4-5]。近年来小分子酪氨酸激酶抑制剂的分子结构、作用机理的研究逐步深入,此类药物在不久的将来定会成为减少和控制癌症发生的有效武器[6],此类药物成为抗肿瘤的热点药物。本文主要研究其中之一盐酸厄洛替尼。
1 合成方法
盐酸厄洛替尼化合物专利CN1137037A于2016年3月到期。我们结合国内外相关专利文献,主要确定了4条合成路线,通过努力摸索,最终确定了我们的生产工艺。根据一致文献和实际研发情况总结如下:
(1)合成路线一
参考化合物专利CN1137037A(申请日:1996.3.28)。

化合物专利收率较高,工艺稳定,原料易得,但是需要用到三氯氧磷等氯代试剂,不利于工业化生产。
(2)合成路线二
参考文献CN1017350157A(申请日:2009.12.30)。
此线路线较长,收率较低,纯化困难,还用到了三氯氧磷,难于工业化生产。
(3)合成路线三
参考文献CN201019102004,CN1137037A(申请日:2010.02.08)。
这条合成路线,步骤长,收率低,用到了三氯氧磷,不适合工业化生产。
(4)合成路线四
参考文献CN102827087A(申请日:2012.6.18)。
此路线收率较高,路线短,原料易得,易于工业化。
我们采用路线4的合成方法,与其它路线相比,路线4没用氯代试剂,且路线短,收率高,原料易得,易于工业化。
2 工艺路线的选择
改进的工艺包括如下步骤:
(1)还原工序,该步反应所使用的反应试剂和专利的一样,反应后处理存在差异,专利通过乙酸乙酯萃取得到中间体1(ErM1),我们根据中间体1的性质通过碱性树脂调节pH后直接过滤得到产品。相比萃取,我们工艺操作更简单,适合工业化生产。
(2)关环工序,该步反应所使用的反应试剂和专利的一样,但是后处理存在差异,专利通过乙酸乙酯萃取得到中间体3,我们根据中间体3的性质结合生成工艺通过过滤得到产品。专利未提到精致中间体3,我们用乙腈∶丙酮(1∶2)精致得到了单杂小于0.1%的中间体3。
(3)成盐工序,专利用二氯甲烷或者乙腈为溶剂成盐,我们用毒性较小的甲醇∶乙醇(1∶1)为成盐溶剂。
(4)精致工序,该步反应所使用的反应试剂和专利的一样,我们通过进一步减少溶剂用量提高了收率。
3 实 验
3.1 主要检测试剂和仪器[7]
高效液相色谱仪(色谱柱:Waters C18(250 mm×4.6 mm,5 μm);流动相:甲醇-pH 3.5磷酸盐缓冲液(20∶80,V/V);流速1.0 mL·min-1;检测波长343 nm;柱温30 ℃;GC惠普1890II;Bruker ARX-300型核磁共振仪(TMS内标)。
3.2 合成工序
3.2.1 还原工序,ErM1的合成
将4.00 kg ErSM1和60.0 kg水加入反应釜,搅拌20 min。控制反应温度(40±5)℃,5 h内分10批加入保险粉。加料完毕,(45±5)℃继续反应,TLC监控反应进程。大约8 h后,反应完全,停止反应。
升温到(60±5)℃,并滴加盐酸。加完后,保温反应2 h,后冷却到(20±5)℃,用碱性树脂调pH=8~9,过滤树脂,搅拌析晶2 h后过滤,用纯化水3.0 kg×2洗。湿品在(60±5)℃下鼓风干燥,得到粗品357 kg。
3.57 kg粗品加入到5.0 kg乙酸乙酯中,加热回流溶解,过滤。向滤液中加入40 kg正己烷,在(20±5)℃搅拌析晶,过滤,滤饼(60±5)℃下鼓风干燥得到ErM1 3.34 kg。收率:83.5%,纯度75%以上。
3.2.2 ErM2的合成工序
3.34 kg ErM1、2.09 kg DMF-DMA和4.68 kg DMF加入到20 L反应瓶中,加热回流,3 h后停止反应。减压蒸馏得到油状物3.12 kg,未进一步处理直接用于下步反应。
3.2.3 关环工序,ErM3的合成
3.12 kg ErM2、1.03 kg间氨基苯乙炔和6.61 kg HAC先后加入到20 L反应瓶中,加热回流,待反应完全(TLC)后停止加热。
冷却到(20±5)℃搅拌下,将反应液倒入50.0 kg冰水中,后加入20.0 kg乙酸乙酯。在(20±5)℃搅拌下,用碱性树脂调节pH=8~9,过滤树脂,有大量固体析出,搅拌2 h后过滤,滤饼60 ℃鼓风干燥得到3.06 kg粗品。
3.06 kg粗品加入到4.50 kg乙腈∶丙酮(1∶2)中,加热回流,大部分溶解后,在(20±5)℃缓慢降温静置析晶(6 h)。过滤,滤饼在60 ℃下鼓风干燥得到2.90 kg(估计值,以实际为准)。
2.90 kg产品加入到4.0 kg乙腈∶丙酮(1∶2)中,加热回流,大部分溶解后,在(20±5)℃缓慢降温静置析晶(6 h)。过滤,滤饼在(60±5)℃下鼓风干燥得到2.75 kg(估计值,以实际为准)。
2.75 kg产品加入到10 kg乙酸乙酯中,加热回流,溶解后过滤,滤液(0±5)℃下析晶(大约2 h),过滤,滤饼60 ℃下鼓风干燥得到2.56 kg(估计值,以实际为准)。
产品单杂若大于0.1%重复用乙腈∶丙酮(1∶2)重结晶过程。
3.2.4 成盐工序,Er的合成
2.56 kg ErM3加入到20.0 kg甲醇∶乙醇(1∶1)中,加热回流至溶液澄清,后冷却到(20±5)℃,酸性阳离子树脂至pH=1~2,过滤树脂,搅拌析晶,过滤并用3.0 kg甲醇∶乙醇(1∶1)洗。滤饼60 ℃鼓风干燥得到2.38 kg盐酸厄洛替尼(粗品)。
3.2.5 精制工序,Er的精制
2.38 kg盐酸厄洛替尼粗品加入到100 L反应釜中,接着加入15.0 kg水和24.0 kg乙腈,加热回流,溶液澄清后过滤,滤液慢慢降温到(20±5)℃,后置于0~5 ℃下进一步析晶。过滤,滤饼(60±5)℃鼓风干燥,得到2.20 kg成品盐酸厄洛替尼。
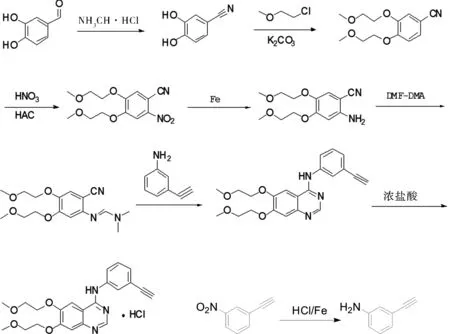
图1 盐酸厄洛替尼的合成Fig.1 Synthesis of Erlotinib Hydrochloride
高效液相色谱仪(色谱柱:Waters C18(250 mm×4.6 mm,5 μm);流动相:甲醇-pH 3.5磷酸盐缓冲液(20∶80,V/V);流速1.0 mL·min-1;检测波长343 nm;柱温30 ℃。测试含量99.97%,水分0.29%,质量收率55%。盐酸厄洛替尼结构示意图如图2所示,1H-NMR数据如表1所示。

表1 H-H COSYTable 1 Data of 1H-NMR

图2 盐酸厄洛替尼的结构Fig.2 The structure of Erlotinib Hydrochloride
(1)11.518 ppmm为单峰,为1号位上的活泼氢。
(2)δ 8.835 ppmm、δ 8.430 ppmm、δ 7.875 ppmm都是单峰芳香氢,即只能是2、3、4、8位碳原子上的氢中,在这三个碳原子上的氢中2上氢的位移最大,其次是3号碳原子上的氢,接着4号 碳原子上氢的位移,最后是8号碳原子上的氢的位移。因此δ 8.835 ppmm 是2位碳原子上的氢,δ 8.430 ppmm是3位碳原子上的氢,δ 7.875 ppmm是4位碳原子上的氢,而11号碳原子上氢的位移包含在了δ 7.397~7.405 ppmm中。
(3)δ 7.776~7.792 ppmm和δ 7.397~7.405 ppmm都是双峰芳香氢,在H-HCOSY谱中都与δ 7.468~7.499 ppmm相关,只能是5、7号碳原子上氢的位移,5号碳上氢原子位移大于7号碳原子上氢的位移。因此,δ 7.776~7.792 ppmm是5号碳原子上氢的位移,δ 7.468~7.499 ppmm是7号碳原子上氢的位移。
(4)δ 4.375~4.392 ppmm和δ 4.309~4.326 ppmm为双三重峰亚甲基的峰,即9、10号碳原子上氢的峰。在H-HCOSY谱中与δ 3.763~3.780 ppmm相关。
(5)δ 4.267 ppmm为单峰,H-HCOSY谱中没有相关峰,因此为11号碳原子上氢的峰。
(6)δ 3.763~3.780 ppmm为三重峰亚甲基的峰,是12、13号碳原子上氢的峰。在H-HCOSY谱中与δ 4.375~4.392 ppmm和δ 4.309~4.326 ppmm相关。
(7)δ 3.322~3.350 ppmm为二重峰甲基的峰,是14、15号碳原子上氢的峰。
从1H-NMR和H-HCOSY图谱可知:精制品各质子化学位移以及裂分情况与盐酸厄洛替尼化学结构相符。
4 结果与讨论
(1)在ErM1的制备过程中,通过用碱性树脂调pH=8~9,过滤树脂,搅拌析晶2 h后过滤得到产品。还原工序,该步反应所使用的反应试剂和专利的一样,反应后处理存在差异,专利通过乙酸乙酯萃取得到中间体1(ErM1),我们根据中间体1的性质通过碱性树脂调节pH后直接过滤得到产品。相比萃取,我们工艺操作更简单,减少有机溶剂的使用,碱性树脂还可回收利用降低了生产成本。
(2)关环工序,通过在(20±5)℃搅拌下,用碱性树脂调节pH=8~9,过滤树脂,有大量固体析出,搅拌2 h后过滤得到产品,再用乙腈∶丙酮(1∶2)重结晶,产品单杂小。该步反应所使用的反应试剂和专利的一样,但是后处理存在差异,专利通过乙酸乙酯萃取得到中间体3,我们根据中间体3的性质结合生成工艺通过过滤得到产品。专利未提到精致中间体3,我们用乙腈∶丙酮(1∶2)精致得到了单杂小于0.1%的中间体3,用碱性树脂调节pH=8~9,过滤树脂,减少有机溶剂使用。
(3)成盐工序,通过酸性阳离子树脂至pH=1~2,过滤树脂,搅拌析晶,过滤并用3.0 kg甲醇∶乙醇(1∶1)洗涤,过滤得产品。专利用二氯甲烷或者乙腈为溶剂成盐,我们用毒性较小的甲醇∶乙醇(1∶1)为成盐溶剂,酸性阳离子树脂至pH=1~2,减少有机溶剂使用。
(4)精致工序,只用到乙腈作为溶剂,用量减少。该步反应所使用的反应试剂和专利的一样,我们通过进一步减少溶剂用量提高了收率。经检验各项指标均符合2020版药典质量标准。
5 结 论
(1)最佳条件确定为:ErM1的制备过程中通过碱性树脂调节pH后直接过滤得到产品,ErM3的合成工序;关环工序,我们用乙腈∶丙酮(1∶2)精致得到了单杂小于0.1%的中间体3;成盐工序,我们用毒性较小的甲醇∶乙醇(1∶1)为成盐溶剂;
(2)测试含量99.97%,水分0.29%,质量收率65%;
(3)通过这次的工艺优化和3批生产验证,表明该工艺适合规模化生产,反应条件可控,经检验各项指标均符合2020年版药典质量标准。
