e1a3 型BCR-ABL伴IKZF1基因突变的成人急性淋巴细胞白血病一例
2023-09-04岑美芝高陆邵梅任明强
岑美芝,高陆,邵梅,任明强
遵义医科大学附属医院血液内科,贵州 遵义 563000
Ph染色体在成人急性淋巴细胞白血病(ALL)患者中阳性率占25%~30%,是9号与22号染色体异位而产生的异常染色体,其产物为BCR-ABL1融合基因。常见的BCR-ABL1 融合基因类型为p190 (典型的e1a2)、p210(e13a2 或e14a2)、p230 (e19a2),而e1a3 为BCR-ABL1融合基因中的罕见变异,鲜有报道[1]。IKZF1是淋巴系统发育必需的转录因子,具有调控淋巴细胞分化的作用[2]。本文报道伴e1a3型BCR-ABL1融合基因且合并IKZF1 基因突变的成人急性淋巴细胞白血病1 例,并复习其相关文献。
1 病例简介
患者女性,59岁,于2021年11月5日因“双下肢乏力1周”入院。1 周前,无明显诱因出现双下肢乏力,于活动后明显加重,无头昏、头痛,无胸闷、气促等不适,今为求诊治就诊我院急诊,查血常规示:白细胞总数10.63×109/L,原幼细胞0.45,血红蛋白82 g/L,血小板5×109/L,以“血小板减少”收入我科。患病以来,精神、饮食尚可,大小便如常,体质量无明显增减。查体:体温(T)36.4℃,心率(P) 78次/min,呼吸(R) 20次/min,血压(BP)121/71 mmHg (1 mmHg=0.133 kPa)。神志清楚,贫血貌,睑结膜稍苍白,四肢可见大片淤斑。辅查:骨髓象,原幼稚淋巴细胞比例占95.0%;免疫分型示在CD45/SSC点图上,可见异常细胞群P2,占有核细胞74.19%,主要表达CD10、HLA-DR、CD34、CD19,部分表达CD13、CD33、cCD79a,弱表达CD20,不表达CD2、CD16、CD7、CD56、CD4、CD41a、CD11b、CD117、CD36、CD64、CD14、CD15、CD11c、CD123、cCD3。在CD45/SSC点图上可见中性粒细胞群Gra,占有核细胞8.87%,比例明显减低,CD13/CD16 表达异常。ALL 相关融合基因检查:BCR-ABL1基因(罕见型)阳性(e1a3)(图1、图2);ALL相关46种基因突变分析:IKZF1基因外显子2~7检测到杂合缺失,为突变异构体(图3)。
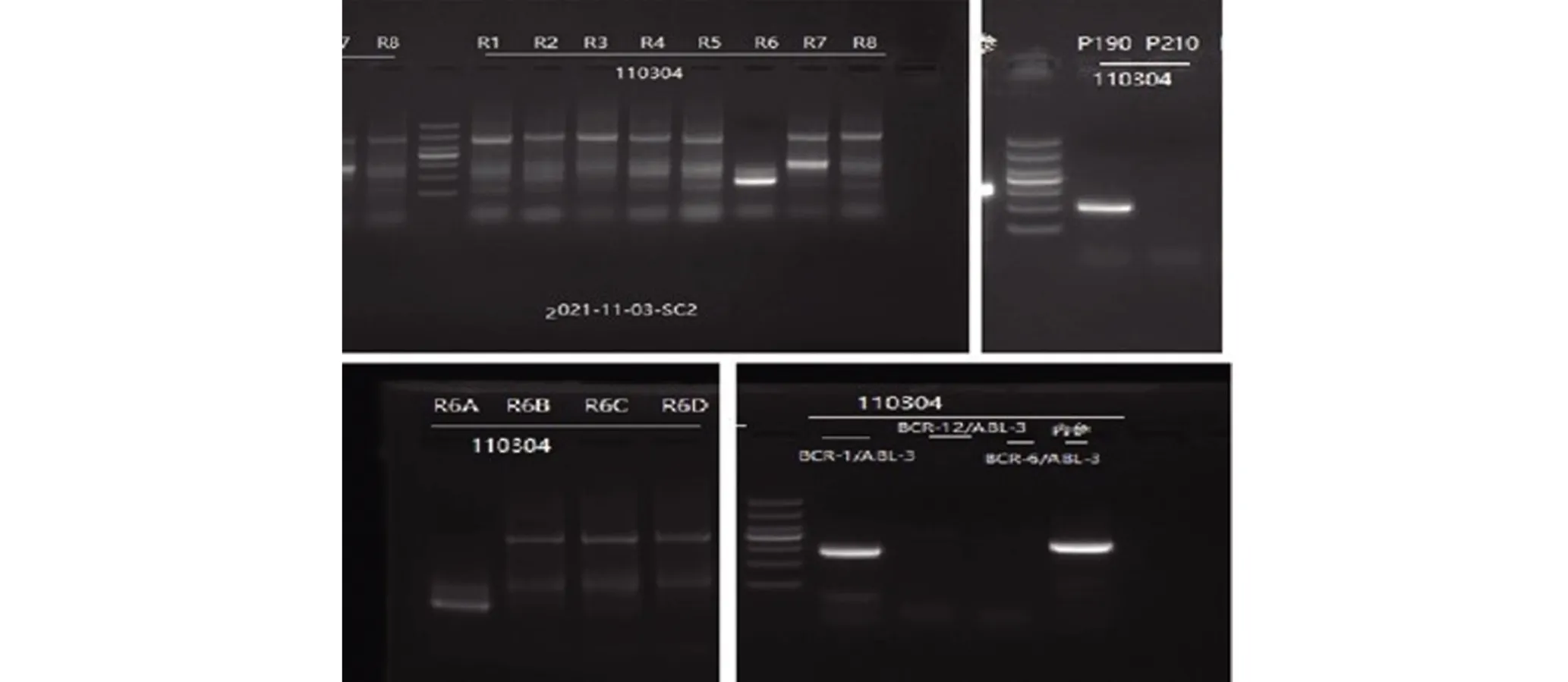
图1 RT-PCR法检测BCR-ABL1融合基因电泳结果Figure 1 RT-PCR results of BCR-ABL1 fusion gene electrophoresis

图2 成人急性淋巴细胞白血病患者BCR-ABL1融合基因扩增产物测序结果Figure 2 Sequencing results of amplified BCR-ABL1 fusion gene in adult acute lymphoblastic leukemia

图3 MLPA法检测IKZF1基因外显子1~8结果Figure 3 Results of exon 1-8 of IKZF1 gene by MLPA
结合病史及上述检查结果,明确诊断为成人急性淋巴细胞白血病(e1a3 型BCR-ABL 伴IKZF1 基因突变)。治疗上予DVP 化疗方案联合甲硫酸伊马替尼胶囊靶向治疗,一般情况好转出院。4 个月后返院复查血常规:白细胞4.42×109/L,红细胞总数3.76×1012/L,血红蛋白113.0 g/L,血小板总数223×109/L。骨髓象:本次骨髓象三系增生可,分类未见幼稚淋巴细胞。BCR-ABL1 融合基因检测为阴性。拟行巩固维持治疗,但患者因经济原因拒绝继续治疗。发病至今9 个月,患者目前生存状态良好。
2 讨论
ALL 是造血干细胞的一种恶性克隆性肿瘤性疾病,来源于不同恶性克隆的各亚型存在显著不同的生物学特征及预后因素,疗效差别很大。
本病例中BCR-ABL1融合基因为e1a3易位,即22号染色体上BCR的外显子1与9号染色体上ABL1的外显子3之间发生易位,导致编码Src同源(SH)3结构域的一部分的ABL1的外显子2(a2)区域在e1a3BCR-ABL1转录本中缺失[3]。该易位不影响编码ABL激酶的APT结合位点,理论上TKI靶向治疗有效[4],因此,本病例患者采用了TKI类药物靶向+化疗方案治疗。经一个周期诱导治疗后,复查提示患者得到了完全细胞学、分子生物学缓解,表明该患者对TKI类药物敏感。同样,Burmeister等[4]筛查发现5例伴e1a3突变的ALL患者,2例未接受TKI 类药物治疗,在化疗后疾病未得到缓解而死亡;其余3例在接受TKI类药物靶向治疗后,得到了完全细胞学缓解,其中2 例还接受了异基因造血干细胞移植,得到了良好的生存。另外,我国目前报道了4例e1a3 型Ph+ALL 患者[5-7],其中1 例未接受化疗+TKI类药物靶向治疗,在化疗后1个月死亡,另外3例在接受化疗+TKI类药物靶向治疗后均得到了缓解,但不幸的是其中一例在5 个月后复发,出现T315I突变,再诱导未缓解,于发病6 个月后因感染死亡。这些报道提示化疗联合TKI类药物治疗是该类患者较好的选择。
IKZF1基因突变为Ph+ALL较常见的突变基因之一,约占80%[8]。IKZF1 基因定位于7p12.2 染色体上,编码Ikaros蛋白。目前经证实的Ikaros蛋白有13种不同的剪接异构体,在功能缺失亚型中,以IK6最为常见,其次为2 号位点外显子到7号位点外显子缺失所致的IK10[9]。IKZF1 基因突变相关研究主要围绕IK6 亚型展开,大部分提示预后不佳[9-11],而针对IK10亚型的研究相对缺如。另有研究表明,具有IKZF1基因突变的ALL早期治疗反应良好者预后良好,早期治疗反应不佳者预后较没有IKZF1基因突变的病例预后差不多[12]。此外,Martinelli等[9]研究发现在Ph+ALL中,IKZF1缺失与无病生存期短和累积复发率高有关,但两组之间的OS没有差异,与预后之间没有相关性。本病例患者合并IK10亚型,在临床症状出现1周后就诊,并得到积极治疗,疗效可观,考虑为早期治疗反应良好者。
综上所述,e1a3易位的Ph+ALL对TKI药物敏感,化疗联合TKI 药物靶向治疗为可取方案;IKZF1 基因突变所致IK10亚型对Ph+ALL的影响有待探究,但早期治疗反应良好是预后良好的反馈。
