非酒精性脂肪性肝病发展及向肝癌转变过程中的分子调控机理研究进展
2023-08-31卞康坤包玉龙
卞康坤,包玉龙,王 利
(内蒙古医科大学基础医学院,内蒙古 呼和浩特 010110)
脂肪性肝病(下称脂肪肝)是常见的代谢性疾病,主要病理表现为甘油三酯在肝细胞中过度蓄积[1]。目前认为脂肪肝的形成与肥胖、酒精、糖尿病、病毒感染和遗传等因素有关。脂肪肝随病情进展,可发展为脂肪性肝炎、肝硬化甚至肝癌。酒精性脂肪性肝病 (alcoholic fatty liver disease,AFLD)和非酒精性脂肪性肝病(non-alcohol fatty liver disease,NAFLD)是脂肪肝常见的两种形式。
NAFLD 是一种多系统疾病,与中枢肥胖症、糖尿病和代谢综合征等密切相关[2-3],比AFLD 发病原因更复杂,治疗难度更大。此外,虽然NAFLD患者的乙型肝炎病毒(hepatitis B virus,HBV)和丙型肝炎病毒(hepatitis C virus,HCV)感染率有所下降,但肝癌发生率却仍在上升[4]。NAFLD 被定义为在没有过量摄入酒精的情况下,肝脏中的脂肪含量达到5%~10%的一类疾病[5]。胰岛素抵抗、代谢综合征或2 型糖尿病以及PNPLA3 或TM6SF2 基因的遗传变异似乎在NAFLD 的发病过程中发挥重要作用[6]。NAFLD 的形成包括2 个阶段:以简单的脂肪变性为特征的非酒精性脂肪肝 (non-alcoholic fatty liver,NAFL),以及伴随脂肪变性、肝细胞膨胀、小叶炎症和纤维化的非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH)[7-8]。目前对NASH 的创新疗法主要针对4 个方面:一是肝脂肪蓄积,二是氧化应激、炎症和细胞凋亡,三是肠道微生物群和代谢性内毒素血症,四是纤维化[9]。随着NAFLD 进展,部分肝纤维化患者会发展成肝硬化、肝衰竭和肝细胞癌(hepatocellular carcinoma,HCC)[10-11]。
NAFLD 已成为严重的全球健康威胁,但NAFLD 发展及向肝癌转变的分子机制仍不清楚,相关的调控信号通路仍然有待研究。Hippo 信号通路、磷脂酰肌醇3 激酶/蛋白激酶B(phosphatidylinositol 3-kinase/protein kinase B,PI3K/AKT)信号通路、转化生长因子-β(transforming growth factorβ,TGF-β)信 号 通 路、腺 苷 酸 活 化 蛋 白 激 酶(adenosine monophosphate-activated protein kinase,AMPK)信号通路和核因子κB(nuclear factor-κB,NF-κB)信号通路是癌症发展过程中的常见调控通路,并在NAFLD 发展过程中发挥着重要作用。笔者综述了NAFLD 发展进程及向肝癌转变过程中涉及的主要信号通路的调控机理研究进展,以期尽可能在NAFLD 的早期阶段阻止其进一步病变及向肝癌的转变,为临床NAFLD 的靶向治疗提供参考。
1 炎症及纤维化在NAFLD-HCC 过程中的作用
炎症和纤维化是NAFLD 发展为HCC 的关键过程。长期慢性肥胖使机体处于一种低度慢性炎症状态,炎症因子的持续产生及信号转导通路的异常为慢性肝病向肝硬化和肝癌的发展提供了有利的微环境。慢性炎症常常是HCC 起始的必要条件,慢性炎症状态下炎症细胞因子持续释放,可能会促进活性氧(reactive oxygen species,ROS)和一氧化氮活性产物的产生,诱导DNA 损伤导致基因突变,从而使细胞发生恶性转变。有研究表明,肝脏中脂质含量增加会导致线粒体功能下降,为增强呼吸满足细胞所需生物能量,进而诱发氧化应激、胰岛素抵抗等,胰岛素抵抗导致肝新生脂肪生成和脂肪组织功能障碍,并诱发炎性细胞因子分泌,逐渐发展为NASH[12]。
纤维化是一种伤口愈合过程,形成纤维结缔组织以替代正常的实质组织。损伤期间正常或轻度纤维化对于组织修复是必要的,广泛或慢性的纤维化会导致胶原蛋白和纤维在细胞外空间的过度积聚[13-14]。若肝损伤是急性、暂时的,纤维化可逐渐被吸收并恢复为正常的肝脏结构;但若肝损伤是慢性、持续的,细胞外基质(extracellular matrix,ECM)则会持续积累,部分纤维化会形成瘢痕组织代替肝实质,造成不可逆的肝纤维化,最终可发展为肝硬化、肝衰竭甚至肝癌[14]。
炎症通常会诱发在NASH 早期阶段观察到的细胞周围纤维化,瘢痕形成会导致进行性纤维化和随后的肝硬化[13-14]。此外,NASH 诱发的肝癌中有35%~50%发生在肝硬化患者和进行常规癌症筛查之前[15-16]。抑制NAFLD 发展过程的炎症和纤维化是阻止NAFLD 相关肝癌发生的关键。
2 相关信号通路在NAFLD 发展及向肝癌转变过程中的调控作用
研究表明,一些细胞因子及信号通路在NAFLD 发展过程中发挥调控作用,其中,炎症因子IL-6、TNF 以及PI3K/AKT 信号通路、AMPK 信号通路和NF-κB 信号通路中的部分因子参与NAFLD-HCC 过程中肝损伤、炎症、纤维化和肝癌细胞自噬的调控(见图1)。Hippo 途径及TGF-β途径则是重点体现Yes 相关蛋白 (Yes-associated protein,YAP)、转 录 因 子 以 及Smad 蛋 白(small mothers against decapentaplegic protein)家族等对NAFLD-HCC 过程中肝脏脂质积累、炎症、纤维化和肿瘤形成的调控(见图2)。
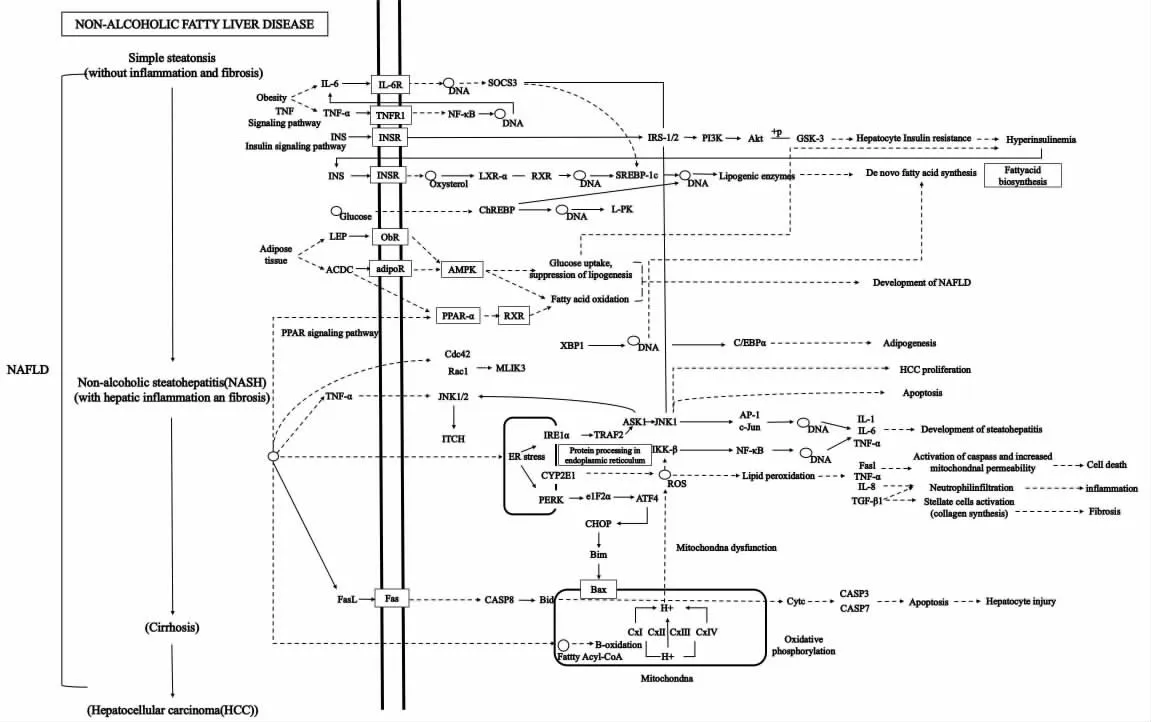
图1 NAFLD-HCC 过程中PI3K/AKT 信号通路、AMPK 信号通路和NF-κB 信号通路参与的炎症因子[17-19]

图2 NAFLD-HCC 过程Hippo 信号通路和TGF-β 信号通路中参与因子[17-19]
2.1 Hippo 信号通路在NAFLD-HCC 过程中调节肝纤维化及肿瘤形成
Hippo 信号通路的主要功能是限制成年人的组织生长并调节发育器官中的细胞增殖分化和迁移,Hippo 途径的失调会导致异常细胞生长和肿瘤形成。大肿瘤抑制因子1/2(large tumor suppressor1/2,LATS1/2)、作为衔接蛋白的salvador 同系物1(salvador homolog 1,SAV1)、YAP 和 具 有PDZ 结合基序的转录共激活因子 (transcriptional coactivator with PDZ-binding motif,TAZ)是该通路的主要信号因子。当YAP 和TAZ 活化时,它们易位到细胞核中以结合转录增强子相关域(transcriptional enhancer associate domain,TEAD)转录因子家族并诱导细胞增殖、存活和迁移[20-21]。在NAFLD 发展至肝癌的过程中,Hippo 信号通路参与调节肝脂质蓄积、肝细胞损伤和纤维化以及肿瘤形成过程。
已知肝星状细胞 (hepatic stellate cells,HSC)的激活在NASH 纤维化中起关键作用[22]。有研究证明肝细胞TAZ 通过作用于HSC 的印度刺猬因子 (Indian hedgehog,Ihh)的分泌促进NAFLD 进程,加重纤维化。TAZ 与肝脏Ihh 基因内含子1 中的TAZ/TEAD 共有序列相互作用,并且该序列可以以TAZ 依赖性方式驱动基因表达,Ihh 促进HSC 中促纤维化基因的表达从而诱导NASH[23],揭示了Hippo 信号通路的下游因子TAZ 在NAFLD 中的促纤维化作用。脂质代谢在肝稳态中至关重要。大肿瘤抑制因子同源物2 (large tumor suppressor, homolog 2,LATS2)在Hippo 途径中主要通过磷酸化使YAP失活从而发挥作用,而活化的YAP 会进入细胞核调控基因转录。近期有研究证明,与冠状动脉疾病相关的连接蛋白(junctional protein associated with coronary artery disease,JCAD)可由肝细胞中的脂肪酸超负荷诱导,在NASH-癌前病变的小鼠模型和人NASH-HCC 标本中高表达,在HCC 中强制过表达可促进肿瘤的生长和增殖。在机制上JCAD与抑癌激酶LATS2 的激酶结构域相互作用抑制LATS2 在Hippo 途径中磷酸化YAP 的能力,YAP的去磷酸化促进了YAP 核易位并上调YAP/TEAD 缔合,导致肝癌细胞中YAP/TEAD 的转录激活,进而上调肿瘤相关因子CCND1 和GLI2 基因表达,促进肝癌细胞的增殖[24]。Aylon 等[25]研究发现,人类NAFLD 患者中,LATS2 表达降低与甾醇调节元件结合蛋白 (sterol regulatory element binding protein,SREBP)活性增加相关,LATS2 抑癌基因也可不依赖YAP 而是作为SREBP 活性的“守门人”,它的下调导致SREBP 活化,通过扰乱胆固醇和脂质的体内稳态促进脂肪性肝病。
功能失调的脂肪自噬可能是肝脂质代谢失调的一个促成因素[26]。已知溶血磷脂酸(lysophosphatidic acid,LPA)可以调节Hippo 途径[27]。研究发现,白细胞分化抗原36(cluster of differentiation 36,CD36)-神经生长抑制因子B(neurite outgrowth inhibitor B,Nogo-B)-YAP 途径可重新编程氧化低密 度 脂 蛋 白 (oxidized low density lipoprotein,OxLDL)代谢,并诱导与NAFLD 相关的HCC 致癌信号。OxLDL 的主要活性成分溶血磷脂酰胆碱(lysophosphatidylcholine,LPC)可以催化LPA,Nogo-B 与自噬相关基因ATG5 相互作用会促进脂肪吞噬,部分地通过LPA 降低非活性(磷酸化)YAP的蛋白质水平,并增加其下游靶结缔组织生长因子(connective tissue growth factor,CTGF)和富含半胱氨酸的蛋白质61 (cysteine-rich protein 61,CYR61)的转录水平,会激活YAP 信号促进肿瘤发生[28]。这表明Nogo-B 与YAP 关联可重新编程OxLDL 代谢并诱导与NAFLD 相关的HCC 致癌信号。
磷酸酯酶与张力蛋白同源物(phosphatase and tensin homolog,PTEN)是AKT 信号的负调节剂,一些人类癌症与PTEN 基因的突变或下调相关[29]。有试验证明SAV1 可以通过抑制肝细胞脂肪酸摄取等途径防止PTEN 缺乏的肝中肝脂质累积,并且肝细胞SAV1 损失会诱导PTEN 缺陷型肝细胞的损伤和纤维化。PTEN 失活与YAP 激活相结合,可在体外显著增加肝脏祖细胞 (liver progenitor cells,LPC)的增殖并导致大的肿瘤球的形成、诱导同种异体移植中的恶性转化[30-31]。这证明了SAV1可以阻止NAFLD 进展和随后的PTEN 缺陷型肝癌的发生,进一步确定了Hippo 途径与NAFLDHCC 之间的重要联系。
胰岛素受体底物2 (insulin receptor substrate 2,IRS2)/AKT 信号通路在肝脂肪形成中起主要作用,包括通过激活SREBP1c 来上调脂肪酸合成基因[32]。PTEN 的丢失会诱导AKT 激活进而稳定TAZ,SAV1 的丢失也会激活YAP/ TAZ,激活的YAP/TAZ 直接诱导IRS2 转录,引起的IRS2 上调促进了PI3K 的活化,进一步增强了已经由于PTEN 缺失而增加的AKT 活性,AKT 激活的增加进一步稳定了TAZ,最终导致AKT 过度激活和NAFLD 发展[33]。这证明Hippo 信号传导可通过调节IRS2 表达与AKT 信号传导串扰加速NAFLD和肝癌的发生。
2.2 PI3K/AKT 信号通路调节肝损伤及HCC自噬
PI3K/AKT 信号通路参与多种细胞功能的调节并广泛参与细胞的生理过程,包括生长、分化、增殖、凋亡、能量代谢和自噬[34]。PI3K 是细胞内脂质激酶家族的成员,其可将3'-磷脂酰肌醇和磷酸肌醇的羟基磷酸化。AKT 属于哺乳动物中保守的丝氨酸/苏氨酸蛋白激酶家族,充当PI3K 的关键下游靶标,并且是PI3K 途径的中央介体,通过磷酸化许多参与细胞存活、增殖、代谢和运动的底物而发挥多种作用[35]。
PI3K/AKT 信号通路在肝损伤中起着至关重要的作用。Lien 等[36]研究发现一氧化碳预处理可以通过诱导肝脏中PI3K/AKT 途径的活化,抑制炎症因子表达,增强抗炎能力并减少肝脏中炎症因子的活性,从而抑制AKT 下游底物糖原合酶激酶3β(glycogen synthase kinase 3β,GSK3β)的活性,减轻肝细胞损伤程度。
PI3K/AKT 信号传导途径是哺乳动物雷帕霉素靶蛋白 (mammalian target of rapamycin,mTOR)公认的上游调节通路,通过激活mTOR 抑制自噬。自噬不仅可以维持肝脏能量和营养平衡,而且在调节肝脏病理生理过程中也起着重要的作用。最近,越来越多的证据表明,PI3K/AKT/mTOR 介导的自噬在不同的肝损伤中起关键作用,并且可能代表了新的治疗肝损伤的靶标[37]。Huang 等[38]发现胰岛素样生长因子结合蛋白相关蛋白1(insulin-like growth factor binding protein-related protein 1,IGFBPrP1)通过促进PI3K/AKT/mTOR 信号传导途径介导的自噬促进HSC 的激活。进一步研究表明,PI3K/AKT/mTOR 途径是诱导HSCs 自噬的主要机制。
PI3K/AKT/mTOR 介导的自噬可能是HBV 感染后诱发肝癌的重要机制。研究报告称,许多癌细胞异常激活AKT 和PI3K,从而导致mTOR 激活,后者通过磷酸化下游p70 核糖体蛋白S6 激酶(p70 S6 kinase,p70S6K)和真核翻译起始因子4E结合蛋白1(eIF4E binding protein,4EBP1)调节癌细胞增殖,从而激活蛋白质合成[39]。Ye 等[40]通过抑制PI3K/AKT/mTOR 途径激活肝癌细胞的自噬,从而有效诱导肝癌细胞凋亡,抑制肝癌细胞的增殖以及肿瘤的侵袭和迁移。该结果的主要原因可能是通过PI3K/AKT/mTOR 途径过度激活肝癌细胞的自噬,从而导致肝癌细胞的自噬细胞死亡(也称为Ⅱ型程序性细胞死亡)。据报道,抑制PI3K/AKT 信号通路可以减少调节胆固醇、脂肪酸、甘油三酸酯和脂肪细胞分化的重要转录因子-SREBPs的表达并显著改善脂质在小鼠体内的沉积[41]。
胰岛素受体底物/磷脂酰肌醇3-激酶/蛋白激酶B(IRS/PI3K/AKT)信号传导途径是通过胰岛素调节血液中葡萄糖平衡的关键途径[42]。该途径的第一步是磷酸化的胰岛素受体与IRS-1 结合传递胰岛素信号。PI3K 是一种脂质激酶,在调节胰岛素的代谢作用中起关键作用[43]。PI3K 可以通过催化和调节p85、p110 亚基影响葡萄糖和脂质代谢[44-45]。PI3K 是一种特异性磷酸化磷脂酰肌醇脂催化剂,可以与IRS-2 结合以传导胰岛素信号[46]。AKT 是PI3K 的直接下游分子,介导胰岛素的各种生物学作用,例如葡萄糖摄取[47]与脂质代谢[48]。有研究表明,肝脏中的IRS-2 特异性调节胰岛素信号传导,并整合胰岛素受体(insulin receptor,InsR)和胰岛素样生长因子-1 受体 (insulin-like growth factor-1 receptor,IGF-1R)信号传导,从而通过PI3K 与AKT 级联介导胰岛素的合成代谢作用[49-50]。
2.3 TGF-β 信号通路加重纤维化并促进NASH向HCC 转变
TGF-β 信号通路可调节细胞增殖、分化、死亡和迁移,在胚胎发生和成人组织稳态中发挥关键作用[51]。TGF-β 可由HSC 激活后释放,最终被磷酸化的Smad2/3 与Smad4 形成异源二聚体或异源三聚体,转移至细胞核激活纤维化基因 (如COL1A1、COL3A1、ACTA2、TGFB1 等)[52]。因此,在NASH 发展过程中,TGF-β 信号转导主要调控纤维化,并有助于NASH 向HCC 的转变。
在NASH 的发展过程中,肝细胞凋亡激活了肝脏再生反应的异常,包括纤维化和炎症[53-54]。肝组织中的慢性炎症刺激巨噬细胞产生的细胞因子TGF-β,会进一步促进肝细胞凋亡,从而加重NASH[55]。有研究发现v-ets 骨髓成红细胞增多症病毒E26 癌基因同源物1(v-ets avian erythroblastosis virus E26 oncogene homolog 1,Ets-1)可充当TGF-β1 信号转导的正调节剂,加速肝细胞凋亡和NASH 的进程。Ets-1 基因敲低减轻了饮食引起的肝细胞凋亡和NASH,并减少了肝损伤/炎症和纤维化。在TGF-β1 存在的情况下,p-Smad2/3 易位到Ets-1 启动子的结合位点,以上调Ets-1 在原代肝细胞中的表达。此外,Ets-1 直接与p-Smad3 结合,从而防止p-Smad3 的泛素化和蛋白酶体降解,增强TGF-β1/Smad3 信号转导的活性,细胞核中持续高水平的p-Smad3 诱导肝细胞凋亡并最终加速NASH 的进程[56],这表明TGF-β 信号通路在NASH 发展过程中对细胞凋亡、炎症反应及纤维化的调节作用。有研究者发现Smad 磷酸亚型可能作为预测NASH 相关HCC 的重要生物标志物[57]。TGF-β1 型受体和c-Jun N-末端激酶 (c-Jun Nterminal kinase,JNK)差异磷酸化介体Smad3,分别产生2 种不同的磷酸亚型:C 末端磷酸化Smad3(pSmad3C)和接头磷酸化Smad3(pSmad3L)。研究人员用不同纤维化阶段的30 例NASH 患者和作为对照的20 例慢性感染丙型肝炎患者研究Smad3 磷酸化与临床进程关联,结果提示高pSmad3L 水平、低pSmad3C 水平可能有助于NASH发展为HCC。肝损伤后,HSC 经历了从脂肪存储、静态表型到高度活化的肌成纤维细胞表型的激活过程,其特征是ECM 的过量产生,最终导致肝纤维化[58]。有研究发现miR-130a-3p 可能通过TGF-β/Smad信号通路直接靶向TGFBR1 和TGFBR2 基因,作用 于TGFBR1 和TGFBR2 基 因mRNA 的3'-UTR,下调TGFBR1 和TGFBR2 基因表达,从而抑制TGF-β1 下游基因的表达,显著降低HSC 激活标记物α-SMA、Smad2、Smad3 和胶原蛋白沉积标记物MMP-2、MMP-9、Col-1、Col-4 的表达水平,在NASH 的进展中负调控HSC 活化和增殖[59],miR-130a-3p 参与TGF-β 信号转导可能有助于阐明肝纤维化的发病机理。Huang 等[60]研究发现Sestrin3(Sesn3)也可以抑制TGF-β-Smad3 信号传导,是ECM 和肝纤维化的关键调节剂。Sesn3 对TGF-β-Smad 途径的关键控制表现在两方面:一是Sesn3 通过与Smad7 的相互作用抑制TGF-β受体;二是Sesn3 通过蛋白-蛋白相互作用和胞质保留直接抑制Smad3 功能。Sesn3 基因敲除小鼠会出现以肝脂肪变性、炎症和纤维化为特征的严重NASH 表型;相反,Sesn3 转基因小鼠在很大程度上不受NASH 发展的影响。Takegoshi 等[61]研究发现支链氨基酸 (branched chain amino acid,BCAA)亦可通过抑制TGF-β1 信号传导来抑制促纤维化信号传导和肿瘤发生。BCAA 通过抑制作为转录因子的核因子Y (nuclear factor Y,NFY)和组蛋白乙酰转移酶p300 的表达来抑制TGF-β1受体1(TGF-β1 receptor1,TGF-β1R1)的启动子活性,并且BCAA 对TGF-β1 信号的抑制作用依赖于哺乳动物雷帕霉素靶蛋白复合物1(mammalian target of rapamycin complex 1,mTORC1)活性,提示存在从mTORC1 到TGF-β1 信号的负反馈调节。这证明了BCAA 通过TGF-β 通路在HSC 中诱导抗纤维化作用,防止肝细胞凋亡,并降低HCC 的发生率。抑制TGF-β 信号转导可抑制纤维化,阻碍NASH 进展直至HCC,为治疗NAFLD 提供了新思路。
Hart 等[62]研 究 了 调 节 肝 脏NAFLD 相 关 纤 维化进程的具体免疫学机制,发现极化1 型[白介素10/4(interleukin10/4,IL-10/IL-4)缺陷型]肥胖小鼠因肝脏中干扰素-γ(interferon-γ,IFN-γ)标记增加而对NASH 具有高度抗性;缺乏IFN-γ 的小鼠TGF-β 和IL-13 信 号 因 子 增 加,迅 速 发 展 为NASH。此外,与在不断扩大的脂肪组织中发现的1 型炎症增加和嗜酸性粒细胞明显减少不同,NASH 的进展与小鼠和人类患者活检组织中嗜酸性2 型肝炎的增加有关。因此,Hart 等[62]提出同时抑制TGF-β 和IL-13 信号传导比单独抑制TGFβ 更完全地减弱了纤维化程度;尽管2 型免疫在脂肪组织中维持正常的代谢信号传导,但与肝脏中的TGF-β 协同作用会加剧NAFLD 的进展。
2.4 AMPK 信号通路调控NAFLD 的多个发展阶段
AMPK 是一种在真核细胞中广泛表达的丝氨酸/苏氨酸蛋白激酶,是一种异源三聚体蛋白激酶复合物。肝脏特有的AMPK 激活可以防止饮食引起的肥胖症和肝脂肪变性。抑制AMPK 活性会导致肝脏的病变从单纯性脂肪变性发展到肝细胞死亡,因此,AMPK 的激活可以从多个方面改善NASH[63]。
AMPK 信号通路在糖类及脂肪代谢调节、能量调控方面发挥重要作用[64]。研究证实AMPK 可在多靶点缓解NASH 的病变程度,AMPK 可通过下调mTORC1、抑制肝脏X 受体(liver X receptor,LXR)的转录以及上调过氧化物酶体增殖物活化受体α (peroxisome proliferator-activated receptor α,PPARα)等途径,降低脂肪酸合成或增加脂肪酸氧化[65-66]。AMPK 通过抑制脂肪从头合成和促进脂肪酸氧化来调节脂质代谢[67-68]。这些结果表明持续的AMPK 活化可实质性地重塑脂质代谢并导致体内总肝脂质水平降低。研究表明,AMPK 活化除了可以抑制新生脂肪形成和促进脂肪酸氧化,AMPK 诱导的自噬也可能有助于减少肝脏中脂质的积累,同时也会加重肝损伤和肝纤维化[69-71],这两者均可促进从NASH 过渡到肝硬化和HCC。
AMPK 的活性受能量状态、营养物质利用以及激素和细胞因子水平的影响[63,71]。高蛋白饮食或增加蛋白质摄入量会降低下丘脑和肝脏中的AMPK 磷酸化,同时增加mTOR 磷酸化。氨基酸水平升高,尤其是亮氨酸水平升高,导致AMP/ATP比降低,从而抑制AMPK 活化[72-73]。肝脏特异性AMPK 激活可防止饮食引起的肥胖,这确实可以改善葡萄糖的体内稳态。胰岛素通过增加葡萄糖摄取和氧化以及通过AKT 介导的AMPKα Ser485/491 磷酸化显著降低AMPK 的活性。同时,AMPK激活改善全身胰岛素敏感性的能力可间接降低脂解作用,从而降低肝脏中的游离脂肪酸水平和脂肪 酸 再 酯 化[74-75]。AMPK 直 接 磷 酸 化 并 抑 制SREBP-1 和SREBP-2,从而减少肝脏中的新生脂肪生成和胆固醇合成[76]。此外,mTOR 通过促进SREBPs 的转录和成熟增加脂肪生成[77]。
炎症是从NAFL 演变为NASH 的标志。包括巨噬细胞、嗜中性粒细胞、树突状细胞和T 细胞在内的免疫细胞的募集,以及免疫细胞衍生的细胞因子、趋化因子和类花生酸的产生会导致肝炎[78]。先前的研究表明,AMPK 的激活会降低促炎介质的表达,并减轻不同条件下的炎症[79-81]。组成性活性AMPK 的肝脏特异性表达降低了炎症基因的表达[71]。促炎细胞因子,例如肿瘤坏死因子α(tumor necrosis factor α,TNFα)显示出抑制AMPK 活性的作用。从机理上讲,AMPK 抑制NF-κB 的核定位,从而抑制NF-κB 靶基因的表达。AMPK 激活导致NF-κB 诱导激酶(NF-κB inducing kinase,NIK)降解,引起非典型NF-κB 通路的衰减,该通路在NAFLD 中被异常激活[82-83]。除抑制促炎信号传导外,AMPK 还可以通过其抗氧化功能改善炎症。ROS 在肝炎的发展中起关键作用。AMPK 抑制核苷酸结合寡聚化结构域样受体蛋白3(nucleotidebinding oligomerization domain-like receptor protein 3,NLRP3)炎性小体的活化,从而预防肝炎[71,84]。
此外,AMPK 与肿瘤的发生发展也有密切关系。AMPK 参与乳腺癌、肺癌、肝癌等癌症的发生[85]。肝癌组织p-AMPK 的浓度较癌旁组织明显降低,且AMPK 的磷酸化和肿瘤组织大小呈明显负相关[86]。研究证实p-AMPK 可降低肝脏内三酰甘油(triglyceride,TG)堆积,AMPK 的磷酸化程度与肝癌的进展和预后呈现一定相关性[65]。
2.5 NF-κB 信号通路调控脂肪性肝炎及纤维化进程
长期以来,基于在促炎基因表达中的作用,NF-κB 途径一直被认为是典型的促炎信号传导途径[87]。NF-κB 信号在受损的肝细胞中激活,上调促炎细胞因子TNFα 和IL-6 的基因表达[88]。早期已有证据表明NF-κB 参与NAFLD 进展的发病机制[87];核 转 录 因 子κB 抑 制 蛋 白 激 酶(inhibitor of kappa B kinase,IKK)/NF-κB 信号通路在NAFLD的发展中起着至关重要的作用[89-91]。
巨噬细胞极化是调节炎症反应的重要机制,有研究发现高脂饮食引起的肝脂肪变性和局部促炎反应与Kupffer 细胞的M1 极化密切相关。棕榈酸(palmitic acid,PA)处理可显著诱导NF-κB 信号通路活化,饱和脂肪酸(saturated fatty acid,SFA)可激活NF-κB 信号通路促进巨噬细胞至M1 表型。过氧化物酶体增殖物激活受体γ(peroxisome proliferator activated receptor γ,PPARγ)与NFκBp65 之间的相互作用对NF-κB 信号传导途径产生拮抗作用,可减轻高脂饮食诱导的M1 型Kupffer 细胞极化并改善肝脂肪变性和炎症反应[92],这提示NF-κB 信号通路通过参与巨噬细胞向M1 型极化调控NAFLD 的炎症反应。此外,环氧二十碳三烯酸(epoxy eicosatrienoic acid,EET)可以通过下调巨噬细胞中NF-κB 通路的激活发挥保护作用,减轻蛋氨酸-胆碱缺乏饮食(methionine and choline deficient,MCD)引起的脂肪性肝炎。游离脂肪酸 (free fatty acid,FFA)处理导致活化的NF-κBp65 从细胞质转移到细胞核,而11,12-EET部分阻止了巨噬细胞中NF-κBp65 转移、IκB 磷酸化和NF-κBp65 的活化[93],提示11,12-EET 可通过抑制NF-κB 激活减轻FFA 诱导的炎症。
炎症和胰岛素抵抗的增加有助于NAFLD 的进展[94]。干扰素调节因子3(interferon regulatory factor 3,IRF3)与细胞质中的IKKβ 相互作用可以抑制IKKβ/NF-κB 信号传导,从而减轻肝脏炎症、胰岛素抵抗和肝脂肪变性。IRF3 的干扰素调节因子相关结构域(IRF-associated domain,IAD)部分与IKKβ 的激酶结构域结合,相互作用可能阻碍了IKKβ-Ser181 的磷酸化和激活,并抑制其下游信号传导。敲除IRF3 基因显著促进慢性高脂饮食引起的肝胰岛素抵抗和脂肪变性;而肝脏细胞IRF3 基因过度表达保留了葡萄糖和脂质平衡[89],IRF3 负调节肝脏中IKKβ/NF-κB 信号传导,有利于脂肪变性和炎症。此外,有研究发现,异硫氰酸烯丙酯(allyl isothiocyanate,AITC)可通过激活沉默调节蛋白1 (silencing regulatory protein 1,Sirt1)/AMPK 途径显著改善肝脂质蓄积,并通过体内和体外抑制NF-κB 途径减轻肝炎性反应。AITC 治疗可在很大程度上降低肝脏组织中TNFα、IL-1β和IL-6 基因的mRNA 转录水平,并且下调IKK、IκBα 和p65 磷酸化[95]。AITC 抑制IKK/NF-κB 信号传导途径可减轻炎症反应,AITC 可能是NAFLD 的潜在治疗剂。
NF-κB 和TNFα 之间的正反馈回路可能会加重炎症和肝细胞损伤的程度[88]。Zhang 等[96]研究发现,miR-378 通过靶向Prkag2 基因,降低Sirt1 的活性并促进NF-κB-TNFα 的炎症途径,miR-378的肝特异性表达通过激活TNFα 信号触发了NASH 和纤维化的发展。miR-378 通过作用于NF-κB-TNFα 信号通路促进NAFLD 的炎症和纤维化。含patatin 样磷脂酶域蛋白3 (patatin-like phospholipase domain -containing protein 3,PNPLA3)I148M(rs738409)在NAFLD 的进展中也起关键作用,它的多态性与肝脂肪含量、肝细胞脂肪性肝炎、纤维化和肝硬化有关[97]。此外,有报道称PNPLA3 I148M 参与NAFLD 中NF-κB 相关炎症调节。人PNPLA3 基因启动子中一个推定的NFκB 结合位点被证明是NF-κB 驱动的PNPLA3 基因转录激活和蛋白质/DNA 复合物形成所必需的,其中,蛋白质/DNA 复合物包含NF-κB p65 亚基和p50 亚基。NF-κB 过表达和PA 处理会使NF-κB与人PNPLA3 基因启动子内源性结合;而PNPLA3基因的沉默可以阻止NF-κB 或PA 诱导的TNFα过度表达[98],这表明NF-κB 通过与PNPLA3 作用促进NAFLD 的炎症反应。
3 问题与展望
随着生活水平的提高以及生活方式的转变,居民饮食结构趋向高脂、高热量,肥胖人口日趋增多,脂肪肝的患病率也逐年增加,使肝脏处于一种损伤状态,继而引发如肝纤维化、肝硬化和肝癌等一系列病变。笔者在文中提及的一些可以调节NAFLD 病变过程中脂质代谢、氧化应激、炎症与胰岛素抵抗等过程的重要信号通路及关键因子,了解这些信号通路能够有效阻遏肝脏损伤的加重,维持肝细胞稳态,调节机体进行自我修复,缓解NASH、肝脂肪变性、炎症、肝纤维化、肝硬化甚至肝癌的症状。然而,目前的研究仅仅是部分揭示了相关因子和信号通路在NAFLD 中的作用,其发展变化机制仍未完全阐明,特别是在由肝硬化向肝癌转化过程中的具体信号通路及机理仍有待研究。因此,基于NAFLD 向肝癌转变过程中的诸多问题,仍需要不断探讨并寻找相关信号通路及调控因子,并阐明其作用机制,以进一步了解由NAFLD 向肝癌转变过程中的病理生理变化,尽早发现恶性转变,为临床靶向治疗提供更加精准的方案。
