MOFs衍生复合氧化物载体上Pd组分分散性与NGVs尾气催化净化性能
2023-08-28杨佳豪岳焮羽廖万年刘来君唐富顺
杨佳豪,岳焮羽,廖万年,刘来君,唐富顺
(桂林理工大学化学与生物工程学院,广西 桂林 541004)
当前,天然气燃料汽车(NGVs)是汽油和柴油汽车的重要替代品。NGVs的优势是温室气体和NOx排放量更低,几乎不会形成炭烟等颗粒物。然而,天然气汽车尾气中未燃烧完全的CH4对全球变暖的影响大约是等量CO2的21倍[1]。因此,必须通过适当的催化末端技术有效地消除这种CH4污染物,以满足日益严格的排放法规,例如中国国6、欧盟6和2021—2027年美国第2阶段重型车温室气体排放标准[2-3],其中美国第2阶段重型车温室气体排放标准涵盖了2018—2027年上市的拖车、卡车、大型皮卡车、厢式货车以及所有类型和尺寸的公共汽车的车型。由于CH4的高度化学稳定性,开发高效的CH4排放控制催化剂仍然具有相当大的挑战性。已有的研究表明,负载型Pd基催化剂能有效地降低废气中的CH4浓度,在过去的几十年里引起了广泛的关注[4]。
一般来说,影响Pd基催化剂CH4转化活性的因素很多,如其物理化学性质、制备方法、Pd组分粒子大小、还原性能以及金属-载体相互作用的程度[5-8]。其中影响Pd基催化剂效率的最重要因素之一是Pd粒子大小,它与Pd的化学状态和活性物种的可及性密切相关[9-10]。但,一个长期存在的问题是:在天然气发动机运行工况下,Pd粒子在高温下易长大,并会导致Pd活性中心烧结而形成更大的聚集体,这使得单位催化剂上的活性中心位点减少,因而为满足排放要求,工程上只有通过增加Pd的负载用量来解决问题。因此,为了降低成本,获得更高效的高温分散型Pd基催化剂,通过具有良好热稳定性的材料来提高Pd活性中心的分散稳定性是需要不断进行改进研究的课题。
CeO2因其可以改善载体的热稳定性、增加贵金属的分散性以及在振动条件下拥有足够的储氧量(OSC),而被广泛添加到目前使用的三效催化剂(TWCs)中[11-12],但它的缺点是热稳定性较差。通过引入过渡金属(Ni、Mn、Cu、Co和Zr)来制备双金属复合氧化物从而提高热稳定性并促进NO的还原,在冷起动阶段实现了较低的起燃温度,使Pd基催化剂在TWCs领域引起了广泛的关注[13-15]。
近年来,利用金属有机骨架(MOFs)作为模板制备具有高孔隙率且均匀的纳米结构金属氧化物载体,因其灵活可控的组分特性而受到广泛关注[16-18]。但还没有研究报道将MOFs衍生载体应用于Pd基催化剂上去除NGVs尾气排放的CH4,NO和CO。因此,本研究拟使用1,3,5-苯三甲酸(H3BTC)作为配体来制备微孔结构CeNi-MOFs和CeZr-MOFs材料,并进一步采用两步分解煅烧法制备出具有MOFs材料衍生特性的多孔型CeNiOx和CeZrOx复合金属氧化物以及负载型Pd/CeNiOx和Pd/CeZrOx催化剂,通过对NGVs尾气净化催化活性的考察,探讨构效关系,以期获得MOFs材料衍生复合金属氧化物载体对贵金属催化剂的分散稳定性的影响规律。
1 试验部分
1.1 催化剂制备
参考文献[19]的方法制备双金属MOFs前驱体:将2 mmol Ce(NO3)3,1 mmol Zr(NO3)4和2.0 mmol H3BTC溶于60 mL DMF中,然后转移至100 mL高压反应釜中,加热至100 ℃后保温反应24 h。反应结束后离心得到固体沉淀,并先用乙醇洗涤2次以除去杂质和过量DMF,再转移到100 ℃的真空干燥箱中干燥24 h。合成产物标识为CeZr-MOF。以上述相同摩尔比的Ce(NO3)3,Ni(NO3)2与H3BTC用量和合成条件制备CeNi-MOF。
将合成的CeZr-MOF研磨成粉末,先在100 mL/min的N2(g)流下以5 ℃/min的速率升温至600 ℃,再将材料在600 ℃下保温3 h。待样品冷却至室温后,在O2(g)流下以5 ℃/min升温至600 ℃,并在600 ℃下保温3 h。待样品再次冷却至室温后,获得所需的CeZrOx复合氧化物并将其标记为CZ-M。用同样的方法煅烧得到了CeNiOx复合氧化物并将其标记为CN-M。
通过定量浸渍法负载1%(质量分数,下同)Pd活性组分的催化剂:将一定量的上述复合氧化物载体在室温下搅拌6 h,使其均匀地分散在所需的硝酸钯溶液中,然后在90 ℃水浴下蒸干水分。最后将获得的固体物转移到马弗炉中,在500 ℃下煅烧3 h,冷却后获得Pd负载量为1%的催化剂(分别标记为Pd/CZ-M和Pd/CN-M)。将催化剂在800 ℃的空气中煅烧8 h得到热老化后的催化剂,分别标记为Pd/CZ-M-a和Pd/CN-M-a。
1.2 催化剂的表征
通过精微高博比表面积及孔径分析仪(JW-BK112)测量样品的比表面积和孔径结构。使用波长1.540 56 Å的CuKα射线作为辐射源,在X’Pert3 Powder多功能X射线衍射仪上获得粉末X射线衍射(XRD)图谱,扫速为0.656 5°/s,扫描步长为0.026 26°,扫描范围为5°~80°。采用H2化学吸附法在PCA-1200全自动化学吸附仪上测量Pd组分的分散性:将0.1 g样品在300 ℃的纯H2下还原1.5 h,还原结束后用N2进行吹扫冷却至室温,然后用N2做载气,向样品中脉冲H2,直到热导检测器(TCD)信号稳定。采用O2化学吸附法在PCA-1200全自动化学吸附仪上测量样品的储氧量(OSC):将0.1 g样品在500 ℃的纯H2下还原1.5 h,还原结束后用He吹扫冷却至室温,然后以He为载气,向样品中脉冲O2,直到TCD检测信号稳定。通过JEOLJEM-2100F,在200 kV加速电压下用透射电子显微镜(TEM)对催化剂的形貌进行表征。
1.3 催化剂的活性测试
采用微反固定床装置评价催化剂的催化活性,气体通过质量流量计控制。活性测试之前,将催化剂在300 ℃的纯H2下还原3 h。反应混合气由1 000×10-6CH4,4 600×10-6CO,1 000×10-6NO,2 300×10-6O2组成,并以N2为平衡气。反应混合气流量为250 mL/min,空速(GHSV)为50 000 mL/(g·h)。出口气浓度由FGA10烟气分析仪进行检测。
2 结果与讨论
2.1 催化剂结构
合成的双金属MOFs的X射线衍射图如图1示,与单晶结构拟合的单金属MOFs晶相结构相比较,观察到双金属材料保留了单金属MOFs的尖锐衍射峰。对于CeZr-BTC,在2θ为8.7°,10.6°,13.5°,17.4°,24.7°和34.4°处观察到特征衍射峰。对于CeNi-BTC,在2θ为7.4°,11.3°,17.4°,23.7°和25.4°处观察到特征衍射峰。说明确实合成了相应的双金属MOFs。
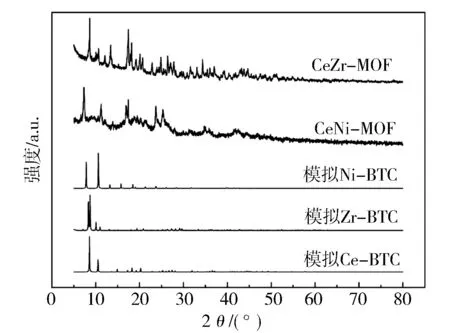
图1 MOFs前驱体的XRD图谱
从图2可见,以双金属MOFs为前驱体衍生制备的未还原状态的负载型Pd/CZ-M和Pd/CN-M催化剂,均可在2θ为28.55°,33.08°,47.47°和56.33°处观察到CeO2的特征衍射峰。这说明,以双金属MOFs为前驱体制备的复合氧化物具有相似的晶相结构并呈现CeO2的晶相特征。同时,与对应的CeO2标准晶相结构(PDF#34-0394)相比,Pd/CZ-M和Pd/CN-M的衍射峰均分别往高角度略偏移0.26°和0.41°,表明Zr和Ni离子均嵌入CeO2的晶格中形成了铈锆和铈镍复合氧化物并使晶格轻微收缩[20]。由此可见,以双金属MOFs为前驱体制备的复合氧化物复合结构更均匀,相互作用更密切。然而,在Pd/CN-M样品上能观察到属于NiO的特征峰,说明在碳化过程中NiO没有完全掺杂到CeO2的晶格中,且在载体表面形成了微小的NiO纳米粒子。与此同时,在两种催化剂上都没有观察到可见的PdO的衍射峰,说明载体表面的PdO粒子太小,无法被X射线衍射检测到。通过Debye-Scherrer公式,以CeO2的2θ=28.55°特征衍射峰计算各个催化剂CeO2的晶体粒子大小,结果如表1所示。Pd/CZ-M和Pd/CN-M的CeO2晶体粒子的平均粒径分别为11.6 nm和7.1 nm,较小的CeO2粒子可以增强Pd-Ce相互作用,有利于提高催化剂的比表面积和Pd组分的分散性[21]。由此可见,以MOFs材料为前驱体来衍生制备复合氧化物时,更易获得结构均一的产物,且有利于形成更细小的Pd粒子,从而提供更多的反应位点,提高催化活性。
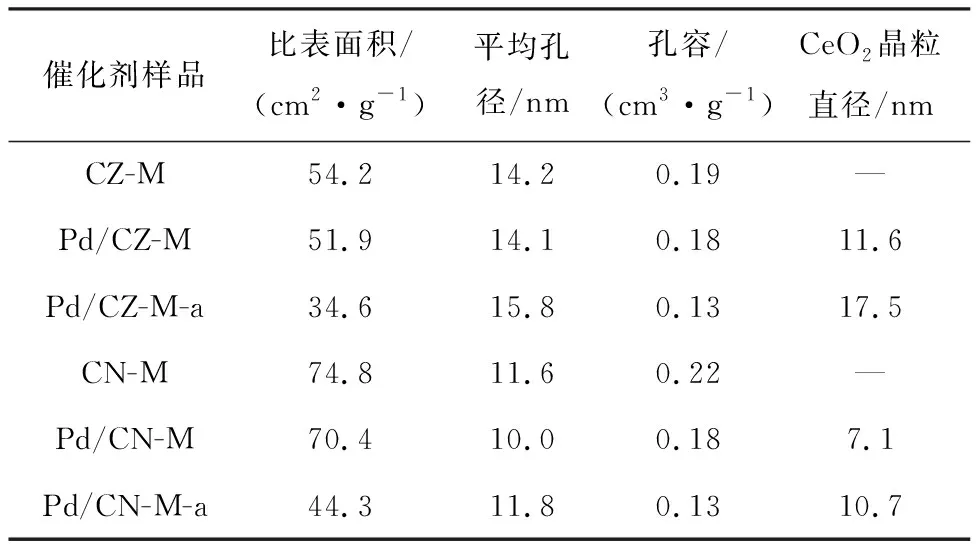
表1 催化剂晶体粒径和孔结构数据
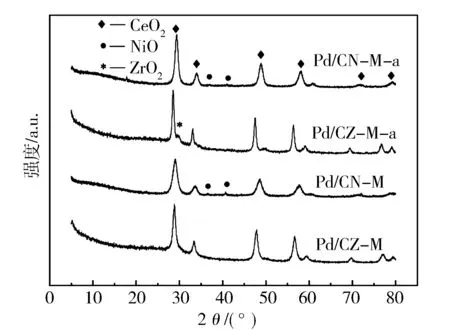
图2 催化剂样品的XRD图谱
未还原状态的催化剂在热老化处理后,Pd/CZ-M-a和Pd/CN-M-a上CeO2的晶体粒径分别增加到17.5 nm和10.7 nm,其中在Pd/CZ-M-a上观察到属于ZrO2的衍射峰(2θ=29.7°),说明CZ-M载体在热老化后结构改变,部分ZrO2在高温下析出,但是整体依然保持原有的复合金属氧化物结构。而在Pd/CN-M-a的XRD衍射峰上依然没有观察到明显的NiO的衍射峰,且CeO2晶体粒径增加不多,说明CN-M在老化后依然保持良好的复合金属氧化物结构,从而提高了催化剂热稳定性。由此证明载体在高温煅烧后虽然略有烧结,但依然没有观察到明显的PdO的衍射峰,说明催化剂在热老化之后依然保持较好的Pd粒子高分散性。
图3a和图3c示出了所有MOFs衍生载体和负载型催化剂的吸附-解吸等温线。这些等温线可认为是Ⅳ型,符合基于IUPAC分类的不规则介孔材料的特性,并且每个样品中都出现E型磁滞回线,表明存在墨水瓶型孔隙。图3b示出了所有载体在2~50 nm范围内的BJH孔径分布图,最可几分布值均在2~4 nm处,这可能是由于MOFs衍生载体是通过两步热解方法制备的:首先,有机配体在N2的保护下分解成碳;然后,由于O2的存在,碳源分解生成气泡,从而转化为大量纳米孔,为Pd的分散提供了有利条件。
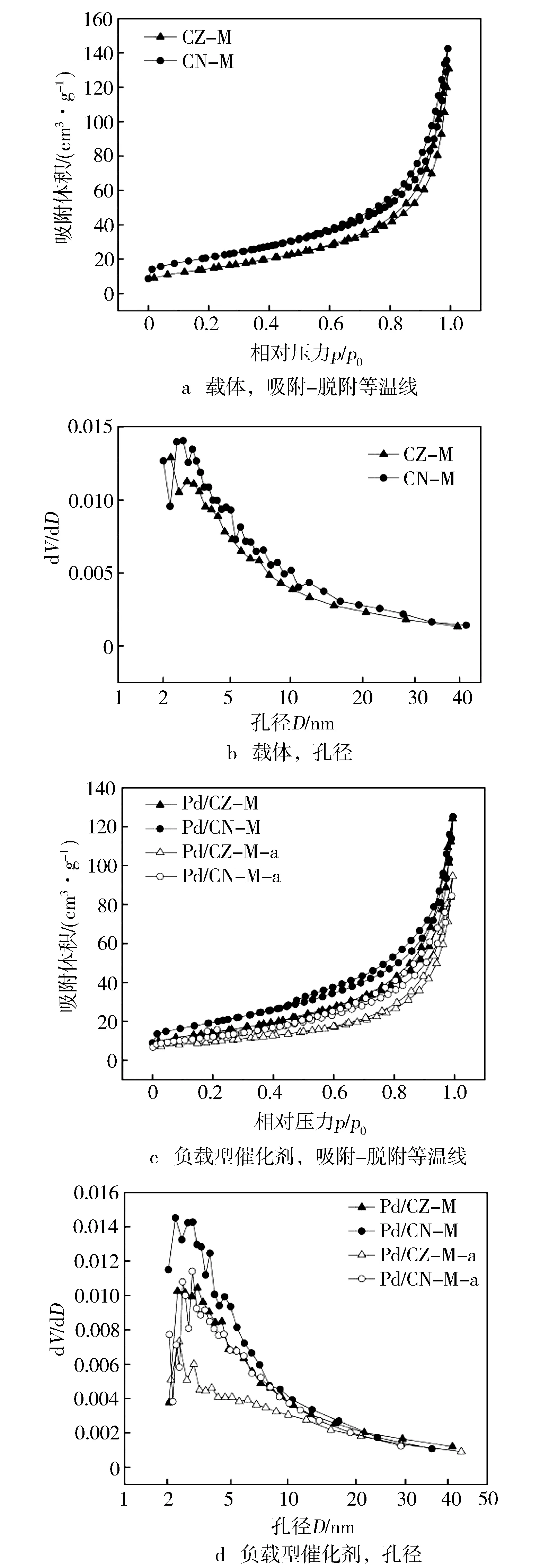
图3 载体与负载型催化剂的N2吸附-脱附等温线和孔径分布图
此外,图3d和表1显示了所有催化剂的孔径分布图和总孔体积,Pd/CZ-M和Pd/CN-M的孔径主要集中在2~5 nm,且总孔体积均为0.18 cm3/g,说明两种催化剂都具有丰富的介孔结构,为Pd的分散提供了有利的条件。对比表1中载体和负载型催化剂的比表面积和总孔体积发现,负载Pd后比表面积和总孔体积都略有减小,说明Pd粒子有可能扩散至载体的孔道中,从而被限制在载体的孔道内形成更细小的Pd粒子。经热老化后,Pd/CZ-M和Pd/CN-M的总孔体积均减少到0.13 cm3/g,且2~5 nm范围内的孔所占的比例减少,说明热老化造成了一些微孔的收缩塌陷,但是总体依然保持良好的孔隙结构。由此可以推测,使用MOFs衍生的复合氧化物作为载体,在热老化后会优先牺牲部分纳米微孔,从而保持整体结构的完整性,以此提高催化剂的热稳定性。
2.2 催化剂上Pd粒子的分散性及储氧量分析
载体和催化剂的储氧量见表2。负载Pd后,Pd/CZ-M和Pd/CN-M的储氧量明显增大。这说明分散良好的Pd物种在氧化还原循环过程中可以吸收更多的O2,由此推测分散良好的Pd物种可能会增加催化剂表面的氧空位,从而增加催化剂的储氧能力,并促进催化氧化反应的进行。经热老化后,催化剂的储氧量均有一定程度的下降,这可能主要归因于催化剂载体和Pd组分的烧结。此外,衍生所得CeNiOx复合氧化物及其负载Pd/CN-M催化剂的储氧量均比相应的CeZrOx及Pd/CZ-M的储氧量高。
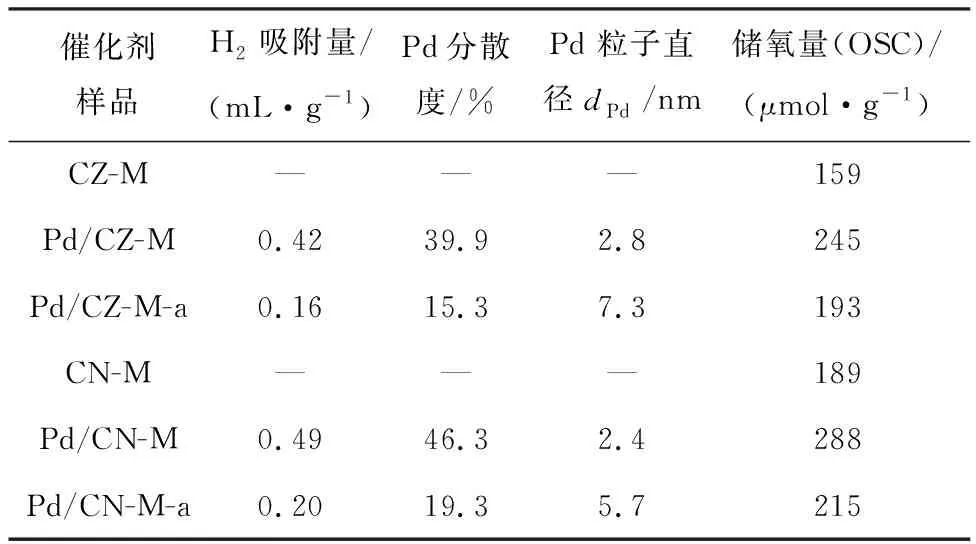
表2 Pd组分分散性和催化剂的储氧量
通过H2吸附测量的Pd在催化剂表面上的分散度和平均Pd纳米粒子尺寸见表2,其中在测量和计算过程中扣除了载体的影响。Pd/CN-M和Pd/CZ-M催化剂的Pd分散度均达到40%及以上,表现出较高的分散特性。这可能是由于催化剂中Pd-Ce间的强烈相互作用可以锚定和稳定贵金属原子,并与Ce4+氧化物的独特氧化还原性能相结合,提高了催化活性。此外,MOFs衍生载体的独特结构和丰富的孔结构也为Pd分散提供了有利条件。
假设Pd粒子为球形,根据由H2吸附结果得到的贵金属Pd分散度可以粗略地按下式估算平均Pd粒子直径[20],计算结果见表2。
式中:ρPd为Pd密度(12 g/cm3);MPd为Pd摩尔质量(106.4 g/mol);SPd为Pd金属摩尔表面积(47.78 m2/mol);DPd为分散度。
Pd/CZ-M和Pd/CN-M催化剂上Pd物种粒径估算值分别为2.8 nm和2.4 nm,这也说明通过MOFs衍生法得到的复合金属氧化物载体的丰富的孔道能对Pd纳米粒子起到锚定和限制作用,可得到更细小且分散更均匀的Pd纳米粒子。而热老化后测量计算得到Pd/CZ-M-a和Pd/CN-M-a的分散度分别为15.3%和19.3%,通过分散度计算得到Pd粒子平均粒径分别为7.3 nm和5.7 nm。由此可以推测载体粒子在热老化过程中团聚长大,孔道发生收缩,同时Pd与载体间的相互作用减弱导致Pd粒子的分散性下降。
2.3 模拟NGVs尾气催化活性
催化剂的CO,NO和CH4转化如图4所示。可以看到,Pd/CZ-M和Pd/CN-M催化剂的转化率曲线都表现出类似的趋势:CO的转化率呈现先增加后减小的趋势;NO转化率先增加后下降再增加直至转化完全;CH4在低温部分几乎没有转化,直到300 ℃以上才开始参与反应,但在350~400 ℃有一个下降过程,随后转化率迅速增加直至完全转化,这表明CH4需在较高的反应温度下才能去除,低温或低速工况,尤其是冷起动时较难转化净化。由于NO2和N2O的生成浓度较低,所以NOx的转化主要表现为NO还原为N2的反应。从CO,NO和CH4的转化特性分析可知,在300 ℃以下的低温反应阶段,由于CH4几乎没有转化,因而NO转化率的增加来源于CO与NO的反应,同时反应混合气中的O2和CO发生氧化反应,故CO转化率呈现迅速上升的趋势,Pd/CZ-M和Pd/CN-M的CO起燃温度(T50)分别为109 ℃和92 ℃,NO的T50分别为140 ℃和114 ℃,表明催化剂具有良好的CO和NO低温转化性能。当反应温度继续增加到200 ℃以上时,由于CO与O2的氧化竞争反应速率增加,氧化竞争反应消耗的CO增加,使得NO与CO反应的还原剂减少,因而NO的转化率下降。当反应温度高于300 ℃时,CH4还原NO的反应起燃,故而NO的转化率又迅速回升至完全转化,这表明高温下CH4与NO的反应速率较大。当反应温度大于400 ℃时,CH4的氧化反应速率增大,该反应消耗的O2量增加,导致没有足够的O2参与CO的氧化反应,使得CO转化率在高温阶段开始下降,这表明高温时CH4的氧化反应竞争性逐渐大于CO的氧化反应。综上所见,CH4不易被活化,CH4的氧化反应较难进行,NGVs尾气中CH4的转化净化达标存在动力学上的难题,仍需要深入研究降低CH4氧化反应活化能的催化剂,探索新型的催化材料。
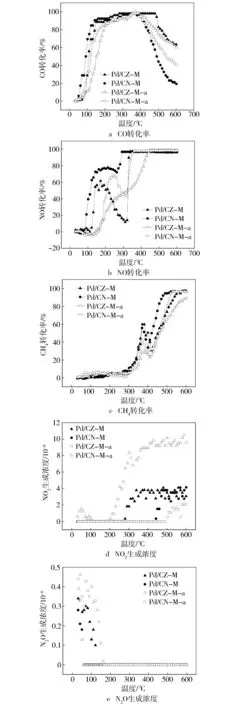
图4 催化剂的CO,NO和CH4 的转化率及NO2和N2O的生成浓度
从图4的活性数据可得到,对于Pd/CZ-M催化剂,CO的T50和达到90%转化率的温度(T90)分别为109 ℃和170 ℃,热老化后则分别往高温方向偏移至152 ℃和210 ℃,而对于Pd/CN-M催化剂,CO的T50和T90分别为92 ℃和120 ℃,热老化后则分别往高温方向偏移至120 ℃和180 ℃。二种催化剂都能在较低的温度实现CO的完全转化且反应速率较快,同时经热老化后T50均只增加大约40 ℃,T90也大约增加40 ℃。对于Pd/CZ-M催化剂,NO的T50和T90分别为140 ℃和330 ℃,热老化后则分别往高温方向偏移至319 ℃和400 ℃,而对于Pd/CN-M催化剂,NO的T50和T90分别为114 ℃和270 ℃,热老化后则分别往高温方向偏移至206 ℃和330 ℃。二种催化剂上NO也能在较低的温度下起燃,且均能在330 ℃左右时达到完全转化,同时经热老化后Pd/CN-M依然保持良好的活性,其T90只增加了大约60 ℃,但是Pd/CZ-M活性下降较为严重,需在400 ℃左右实现NO的完全转化。对于Pd/CZ-M催化剂,CH4的T50和T90分别为350 ℃和480 ℃,热老化后则分别往高温方向偏移至446 ℃和580 ℃,而Pd/CN-M催化剂的T50和T90分别为350 ℃和450 ℃,热老化后则分别往高温方向偏移至370 ℃和480 ℃。由此可见,对于CH4的转化,所有新鲜催化剂在350 ℃均能达到50%以上的转化率。随着反应的进行,转化率略有下降,但是依然能在500 ℃左右实现CH4的完全转化。同时在热老化后催化剂在中高温部分活性都有所下降,其中Pd/CN-M-a依然能在480 ℃左右实现CH4的完全转化,而Pd/CZ-M-a活性虽然下降较多,但是依然能在580 ℃实现CH4的完全转化。
2.4 构效关系探讨
综合前述二种催化剂及老化后的活性数据可见,新鲜催化剂时,Pd/CN-M催化剂的CO,NO和CH4的T50分别比Pd/CZ-M催化剂的低17 ℃,26 ℃和0 ℃,两者相差不多,但其CO,NO和CH4的T90分别比Pd/CZ-M催化剂的低50 ℃,60 ℃和30 ℃,显示出更好的低温活性。当催化剂在空气中经过800 ℃,8 h老化后,Pd/CN-M-a催化剂的CO,NO和CH4的T50分别比Pd/CZ-M-a催化剂的低32 ℃,113 ℃和76 ℃,并且其CO,NO和CH4的T90也分别比Pd/CZ-M催化剂的低30 ℃,70 ℃和100 ℃。这表明,无论是新鲜还是老化后的催化剂,Pd/CeNiOx催化剂模拟NGVs尾气的CO,NO和CH4三效净化催化活性均优于Pd/CeZrOx催化剂,尤其是对NO和CH4的催化转化净化表现出更优良的低温性能。另外,老化后,Pd/CeNiOx催化剂的CO,NO和CH4的T50分别往高温方向偏移28 ℃,92 ℃和20 ℃,T90分别往高温方向偏移60 ℃,60 ℃和30 ℃,而Pd/CeZrOx催化剂CO,NO和CH4的T50分别往高温方向偏移43 ℃,179 ℃和96 ℃,T90分别往高温方向偏移40 ℃,70 ℃和100 ℃,可见Pd/CeNiOx催化剂其热稳定性要高于Pd/CeZrOx催化剂。根据前述的孔结构分析可知,老化后Pd/CeNiOx催化剂的比表面积也比Pd/CeZrOx催化剂的更大,微孔的收缩塌陷更小,更利于Pd物种的分散。从H2吸附和OSC表征分析结果可得到,Pd/CN-M催化剂上老化前和老化后的Pd物种粒子直径分别为2.4 nm和5.7 nm,均比Pd/CZ-M的2.8 nm和7.3 nm小,尤其是老化后Pd/CeNiOx催化剂上Pd物种粒子稳定性更佳,催化剂的储氧量也更高。正是这种更高的分散稳定性,使得Pd/CeNiOx催化剂老化后的热稳定性要高于Pd/CeZrOx催化剂,因而其三效净化催化活性具有更佳的低温活性稳定性,尤其是NO和CH4催化转化性能更佳。
进一步对老化后样品进行高清透射电镜(HRTEM)观察(见图5),得到未还原状态的Pd/CN-M-a和Pd/CZ-M-a催化剂的PdO粒子平均粒径分别为6.0 nm和7.5 nm,相比于以往报道过的一些文献数据[20,22](见表3),如1%Pd/Al2O3-CeO2和1.6%Pd/P-Al2O3催化剂的PdO粒子平均粒径分别为8.5 nm和19.0 nm,本研究以CeNi-MOFs和CeZr-MOFs材料为前驱体碳化衍生并负载得到的Pd/CeNiOx和Pd/CeZrOx催化剂上PdO粒子平均粒径更小,分散性和稳定性更佳。对于催化剂性能来说,老化后的Pd/CN-M-a和Pd/CZ-M-a催化剂CO和NO的T50和T90比1%Pd/Al2O3-CeO2和1.6%Pd/P-Al2O3催化剂的低90~180 ℃,同时CH4的T50和T90也低20~120 ℃。由此可见,通过MOFs衍生的复合金属氧化物载体具有丰富的孔道结构,更强的Pd-Ce间相互作用,可更好地锚定Pd组分,实现Pd的均匀分散,同时双金属MOFs中的过渡金属Zr和Ni掺杂到CeO2的晶格内,增强了载体的热稳定性,从而可适度改善Pd组分的分散热稳定性。分散良好的Pd物种为催化剂提供了更多的可吸附位点和氧空位,增强了催化剂的氧化还原性能,从而改善NGVs三效催化性能,使CO,NO,CH4在更低的温度下完全转化。当然,不同金属节点的MOFs材料碳化衍生得到复合氧化物为载体时对Pd活性中心的分散热稳定性的改善效果不尽相同,以CeNi-MOFs材料碳化衍生并负载得到的1%Pd/CeNiOx催化剂上分散热稳定性改善效果比CeZr-MOFs材料更好,具有更佳的低温活性稳定性,尤其是NO和CH4催化转化性能更佳。从改善的情况来看,Pd粒子的粒径仍偏大,CH4的转化温度仍较高,有待进一步广泛深入地研究。尽管如此,以CeNi-MOFs材料碳化衍生并负载得到的1%Pd/CeNiOx催化剂对活性组分的分散热稳定性改善是有效的,试验结果也为后续的研究提供了有益的经验和认识。
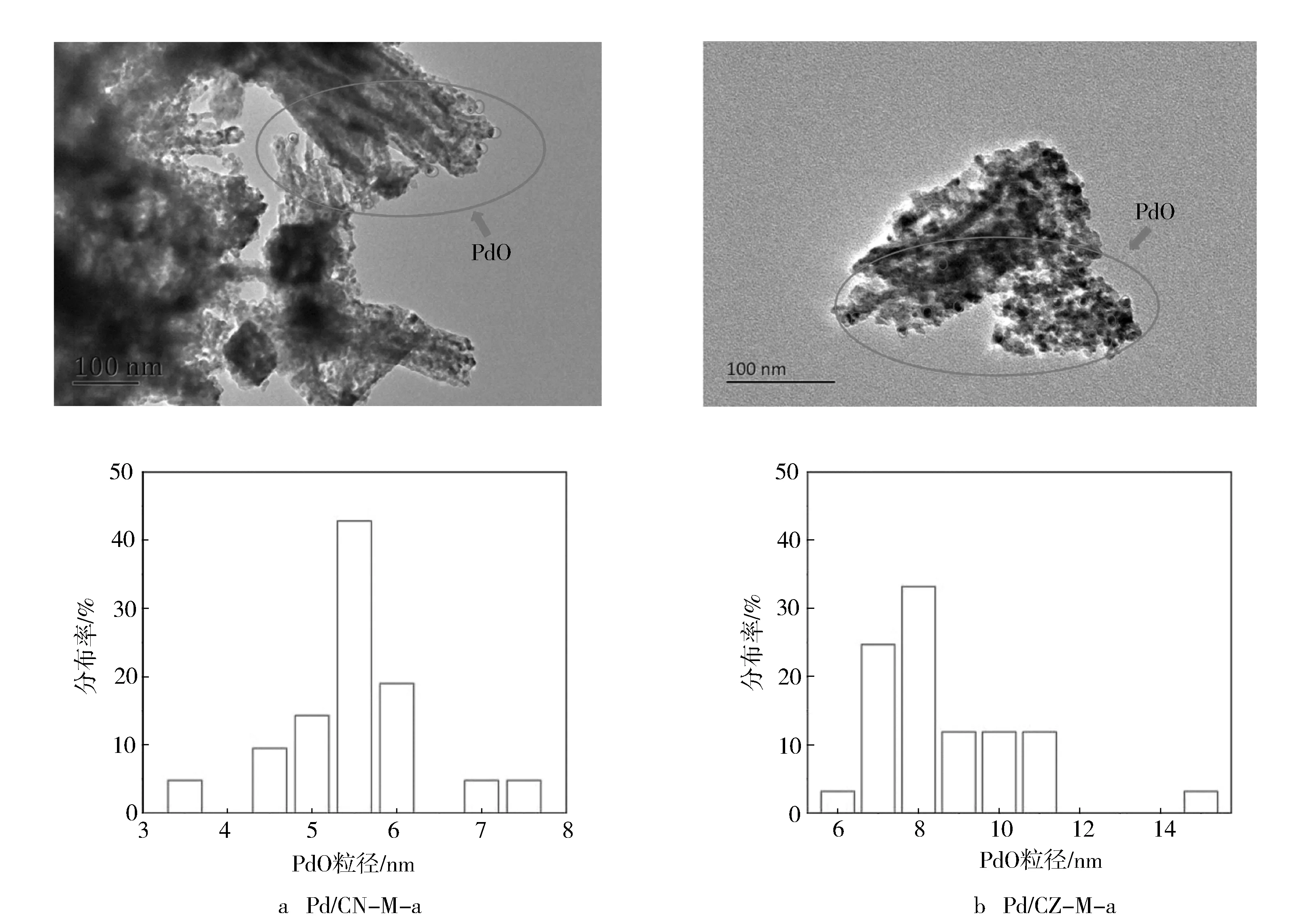
图5 Pd/CN-M-a和Pd/CZ-M-a的HRTEM图谱和粒径分布
3 结束语
相比以CeZr-MOFs材料碳化衍生并负载得到的1%Pd/CeZrOx催化剂,以CeNi-MOFs材料碳化衍生并负载得到的1%Pd/CeNiOx催化剂,其老化后模拟NGVs尾气CO,NO和CH4三效净化催化活性具有更佳的低温性能,尤其是NO和CH4催化转化性能更佳,CO,NO和CH4起燃温度T50和T90也更低。老化后的Pd/CeNiOx催化剂的CO,NO和CH4起燃温度T50分别往高温方向偏移28 ℃,92 ℃和20 ℃,T90分别往高温方向偏移60 ℃,60 ℃和30 ℃,偏移后T50分别为120 ℃,206 ℃和370 ℃,T90分别为180 ℃,330 ℃和480 ℃。
Pd/CeNiOx和Pd/CeZrOx催化剂老化后Pd物种粒子直径分别从2.4 nm和2.8 nm增大到5.7 nm和7.3 nm,相对地Pd/CeNiOx催化剂上Pd物种粒子稳定性更佳,催化剂的储氧量也更高。通过具有丰富孔道结构的MOFs材料来衍生制备复合金属氧化物载体,可以适度改善Pd组分的分散热稳定性,并提供更多的可吸附位点和氧空位,从而改善NGVs三效催化性能。但CH4的转化温度仍较高,需进一步地广泛深入研究。
