基于BSA-seq结合连锁分析发掘大豆荚粒性状QTLs及候选基因
2023-08-26孙亚倩陈士亮褚佳豪李喜焕张彩英
孙亚倩, 陈士亮, 褚佳豪, 李喜焕*, 张彩英*
(1.河北农业大学,华北作物改良与调控国家重点实验室,华北作物种质资源研究与利用教育部重点实验室,河北 保定 071001;2.承德市农林科学院,河北 承德 067000)
大豆是重要粮油作物,提供了全球50%以上的食用油和25%以上的植物蛋白[1]。然而,大豆单产水平和单位面积经济效益较其他农作物而言相对较低,亟需进一步提高以满足日益增长的需求。豆荚和籽粒是菜用和粒用大豆重要的产量相关性状,直接影响单位面积产量和经济效益[2]。荚粒性状还是菜用和粒用大豆最重要的外观品质性状,对其商品价值具有重要影响。因此,研究荚粒性状对提高大豆产量、品质、商品价值和经济效益等具有重要意义。
大豆荚粒性状包括荚长、荚宽、荚重、粒长、粒宽和粒重等,这些性状属于数量性状,且已获得少数控制其遗传位点及候选基因。Niu等[3]获得59个粒长、粒宽和粒厚等性状QTLs,位于20条染色体;Xie 等[4]通过精细定位6 号染色体籽粒大小QTLs获得8个候选基因;Dargahi等[5]获得16个粒长、粒宽和粒厚QTLs,位于9 条染色体,表型贡献率7.4%~18.4%;Yang 等[6]检测到60 个籽粒性状QTLs,位于除5 和19 号以外的18 条染色体;Teng等[7]研究发现,5 个粒长QTLs 位于7、12、13 和17号染色体,3 个粒厚QTLs 位于9、12 和13 号染色体,5 个粒宽QTLs 位于12、13 和17 号染色体;Cui等[8]获得30 个籽粒性状QTLs,位于12 条染色体,包括1 个多环境下的一因多效QTL;Hina 等[9]认为,6、10、13和20号染色体存在籽粒性状QTLs 热点区;Kumawat 等[10]获得53 个籽粒大小QTLs,位于除3、14和18号以外的17条染色体。
目前,随着测序技术的发展,混合群体分离测序(bulked segregant analysis sequencing,BSA-Seq)因无需构建分子遗传连锁图谱,在重要性状遗传位点发掘中越来越受到重视[11‑12]。张之昊等[13]采用BSA-seq 技术挖掘大豆的多小叶基因,经SNPindex 算法获得11号染色体的3个关联区域,总长度3.42 Mb,包含701 个基因;曾维英等[14]采用BSA-seq 技术挖掘大豆抗豆卷叶螟基因,经SNPindex 算法获得329 个基因,位于7、16 和18 号染色体;刁卫楠等[15]通过BSA-seq 技术将控制西瓜黄色果肉的主效QTLs 定位于6 号染色体,并结合基因注释和表达分析获得5 个候选基因;Zhao等[16]利用BSA-seq 方法发掘到水稻3 号染色体控制粒长的基因热点区,并通过基因编辑技术证实了粒长基因;Zhang 等[17]利用BSA-seq 方法获得3 个水稻株高QTLs、2 个穗长QTLs 和4 个抽穗期QTLs。
综上,尽管目前已有关于大豆荚粒性状遗传位点发掘与相关基因研究,但多基于二代分子标记连锁定位方法获得,因图谱密度和饱和度较低,所得连锁标记及基因数目少,且很难在育种实践中应用。鉴于此,本研究基于大豆重组自交系群体(recombinant inbred line, RIL)在多种环境条件下的荚粒性状(荚长、荚宽、荚重、粒长、粒宽和粒重),筛选极端家系并构建2个混池,通过BSA-seq技术发掘控制荚粒性状的遗传位点,结合前期通过连锁定位方法获得的该群体QTLs[18],进一步发掘在多环境条件下、2种方法同时能检测到的一因多效遗传位点;同时结合RIL群体亲本荚粒不同发育时期转录组数据筛选候选基因,为开展大豆荚粒性状分子遗传改良和遗传基础解析提供支撑。
1 材料与方法
1.1 试验材料
供试大豆重组自交系群体(C813×KN7,F6:8,193个家系)[18]由河北农业大学大豆遗传育种课题组提供,其亲本C813为大荚大粒菜用大豆优异种质(鲜荚长 5.0 cm、鲜荚宽1.3 cm、鲜荚重2.2 g、鲜粒长1.4 cm、鲜粒宽0.8 cm、鲜粒重0.6 g),KN7 为小荚小粒育成品种(鲜荚长4.2 cm、鲜荚宽1.1 cm、鲜荚重1.4 g、鲜粒长1.2 cm、鲜粒宽0.8 cm、鲜粒重0.4 g)。
1.2 试验方法
1.2.1大豆重组自交系群体种植 供试大豆重组自交系群体分别于2020、2021 年种植在4 种环境条件下,即2021 年保定(E1)、2020 年保定(E2)、2020 年石家庄鹿泉(E3)、2020 年廊坊文安(E4),田间试验采用完全随机区组设计,行长3 m,行距0.5 m,株距10 cm,3次重复,田间种植和管理参考Chen等[18]方法。
1.2.2大豆重组自交系群体荚粒性状鉴定 待大豆重组自交系群体生长至R6~R7 期,取植株中部生长一致的标准二粒荚10 个,测定其荚长(pod length, PL)、荚宽(pod width, PW)和荚重(pod weight, PWT),重复3 次[18];随后剥去荚皮,将所有籽粒置于SC-G种子自动分析系统(杭州万深检测科技有限公司)样品板上,分析粒长(seed length,SL)、粒宽(seed width, SW)和粒重(seed weight,SWT)[2,18-20]。
1.2.3重组自交系群体荚粒性状极端家系筛选根据供试大豆重组自交系群体各家系在不同环境条件下荚粒性状的综合表现,分别选择荚长>4.5 cm且荚宽>1.2 cm的20个大荚大粒极端家系和荚长<4.5 cm 且荚宽<1.2 cm 的20个小荚小粒极端家系,用于BSA-seq发掘控制大豆荚粒性状QTLs。
1.2.4大豆荚粒性状混池BSA-seq 分别提取上述入选的家系以及RIL群体亲本(C813和KN7)基因组DNA,并将其按照性状分类,等量混合成2个混池——大荚大粒池(large pool,LP)和小荚小粒池(small pool,SP),分别含有20 个家系。将LP、SP、C813 和KN7 的DNA 送广州基迪奥生物科技有限公司完成BSA-seq 分析,测序深度为30×,参考基因组为Williams82 v2.0,测序质量通过Q20(高通量测序中,错误率低于1%的碱基百分比)和Q30(高通量测序中,错误率低于0.1%的碱基百分比)等进行评估。
另外,为保证结果可靠性,按照以下标准筛选SNP 和Indel 标记:①入选标记在亲本间存在差异,且符合RIL 群体分离方式;②RIL 群体亲本测序深度大于阈值;③标记在2 个混池中均不缺失;④每个混池测序深度需大于10×且小于500×;⑤至少1 个混池的SNP 指数(SNP index,SNPI)在0.3~0.7之间。
1.2.5发掘荚粒性状遗传位点的算法 本研究采用3 种算法进行荚粒性状遗传位点发掘,包括SNP index 算法、欧氏距离(Euclidean distance,ED)算法和G-statistic 算法[13,21];置信水平分别设为0.95和0.99,3种算法的计算公式如下。
①SNP index算法。
式中,SNPI为SNP 指数,ρ 为位点在混池中的深度。
②ED算法。
式中,mut与wt为隐性和显性混池,A、C、G、T为各等位标记位点的覆盖深度。
③G-statistic算法。
式中,O为等位深度的观测值,E为等位深度的期望值,ln为自然对数,i为等位基因数目。
1.2.6BSA-seq 一致性区间候选基因筛选 首先,比较供试大豆RIL群体BSA-seq不同算法关联结果,筛选一致性关联区间;然后,在一致性区间内寻找候选基因,并结合基因注释、基因DNA 水平SNP 等位变异以及RNA 水平表达分析,筛选大豆荚粒性状相关基因。候选基因查找及其基因注释参考大豆测序基因组Williams82(Glyma2.0)(https://www.soybase.org/),基因的RNA 水平表达分析采用供试RIL群体亲本C813和KN7的豆荚5个不同发育时期(Pod-T1~Pod-T5)和籽粒3 个不同发育时期(Seed-T1~Seed-T3)的基因表达转录组数据[2]。
1.2.7表型数据分析 利用Microsoft Excel 2017
统计软件的成组数据t检验方法,对供试RIL群体极端家系在单一环境条件下以及多种环境条件下的6 个荚粒相关性状(荚长、荚宽、荚重、粒长、粒宽和粒重)进行显著性检验。
2 结果与分析
2.1 大豆重组自交系群体BSA-seq 极端家系的遴选
通过分析供试大豆重组自交系群体2 年4 种环境条件下的荚长、荚宽、荚重、粒长、粒宽和粒重,筛选出40 份极端家系构成2 个混池——LP 和SP。分析2 个混池不同环境条件下的荚粒性状(表1)发现,2 个混池在单一环境条件下的荚长、荚宽、荚重、粒长、粒宽和粒重均存在极显著差异,并且在多环境条件下的荚粒性状亦存在显著或极显著差异,为进一步利用BSA-seq 方法发掘控制其遗传位点奠定了重要基础。

表1 大豆RIL群体BSA-seq极端家系不同环境荚粒性状遗传差异Table 1 Genetic variation of extreme RIL lines on pod and seed traits for BSA-seq under different environments
2.2 BSA-seq测序质量及SNP与Indel分析
通过分析2 个混池及其亲本材料的BSA-seq测序质量(表2)发现,过滤后的有效数据量为30 Gb,Q20 在97.2%以上,Q30 在92.5%以上,G和C 碱基含量为36.32%~36.58%,过滤后的总读段为2 Gb;与参考基因组的平均比对效率在98%以上,且基因组覆盖度深,4 个样品的10×基因组覆盖率均在94%以上,20×基因组平均覆盖率则在80%以上,说明4 个样品的BSA-seq 测序数据充足且质量高,可进行后续BSA-seq 关联位点分析。

表2 两个混池及其亲本BSA-seq测序质量分析Table 2 Quality analysis of BSA-seq of two pools and soybean RIL parents
进一步分析BSA-seq 测序SNP(表3)与Indel(表4),共获得162万~241万个SNP,其中位于外显子区的有6.2 万~9.1 万个,基因上游的有9.6 万~14.4万个,引起非同义突变的有3.5万~5.1万个,引起提前终止的有726~1 089个,引起终止密码子丢失的有144~223个;同时获得30万~43万个Indel,其中位于外显子区的有3 823~5 338个,基因上游的有3.0 万~4.4 万个,引起移码缺失的有1 128~1 534个,移码插入的有987~1 314个,引起提前终止的有69~95 个,引起终止密码子丢失的有10~18个。

表3 两个混池及其亲本BSA-seq测序SNP数量与质量分析Table 3 SNP number and quality analysis of BSA-seq of two pools and soybean RIL parents
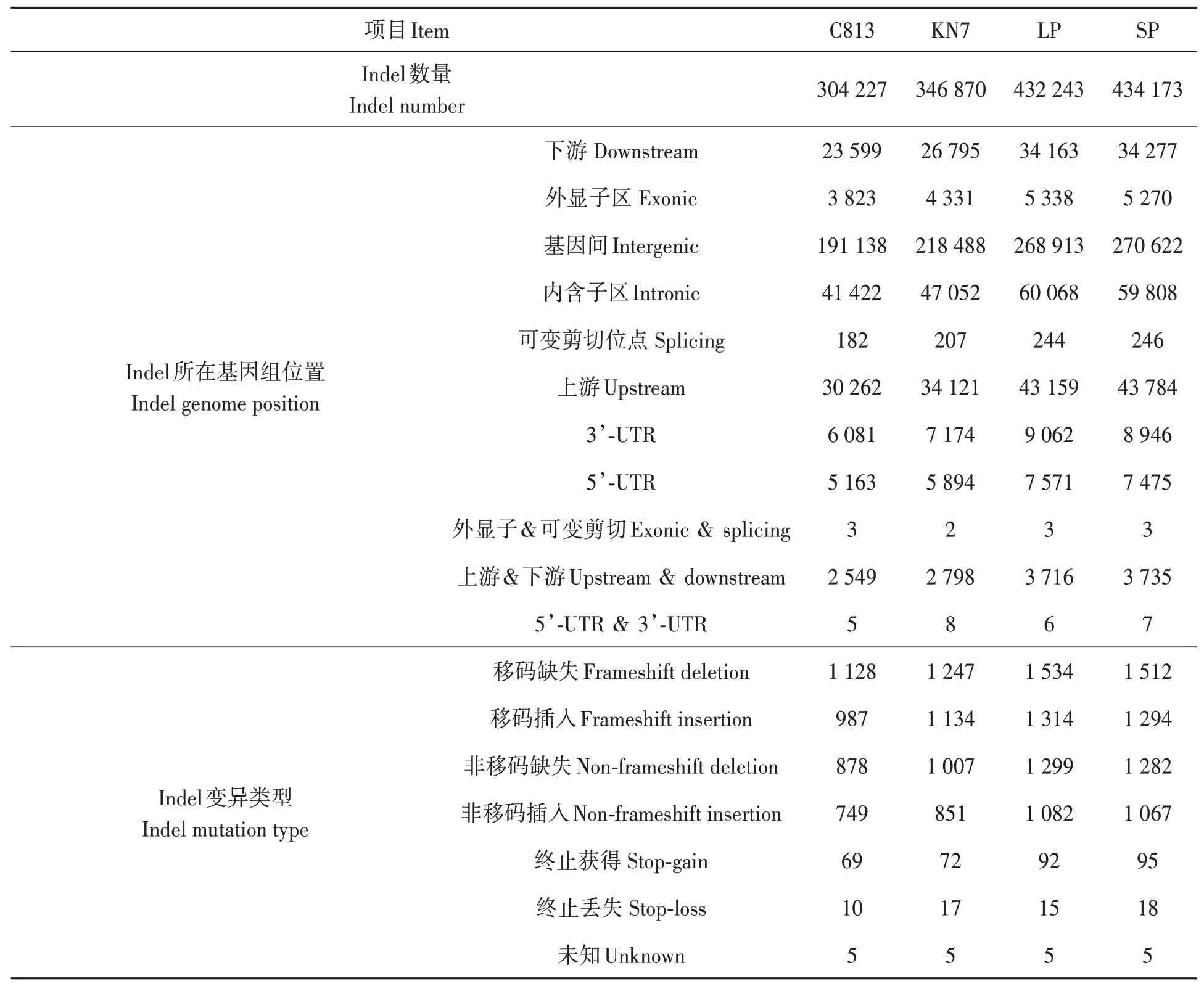
表4 两个混池及其亲本BSA-seq测序Indel数量与质量分析Table 4 Indel number and quality analysis of BSA-seq of two pools and RIL parents
2.3 大豆荚粒性状BSA-seq遗传位点发掘
通过分析筛选后的SNP 和Indel标记发现,符合入选标准的总标记数量为763 727个,其中包括SNP 标记658 186 个、Indel 标记105 541 个,并以17、9和3号染色体入选标记数量最多。在此基础上,利用上述SNP 和Indel 标记分别通过SNPindex、ED 和G-statistic 算法发掘荚粒性状关联位点。
2.3.1SNP-index 算法发掘遗传位点分析 采用SNP-index 算法分析与荚粒性状关联位点,结果(图1A)表明,在置信度为0.95 时,获得27 个关联区域,位于15 条染色体(除5、8、10、11 和18 号外),其中7 号染色体包括7 个区域,16 和20 号染色体各包括3 个区域。同时发现,在置信度为0.99 时,获得5 个关联区域,分别位于5 条染色体(1、4、7、12和19号)。

图1 大豆荚粒性状不同算法关联区域Fig. 1 Associated regions of soybean pod and seed traits via different methods
2.3.2ED算法发掘遗传位点分析 采用ED方法分析与荚粒性状关联的位点,结果(图1B)表明,在置信度0.95 时,获得41 个关联区域,分别位于17 条染色体(除5、8 和11 号外),其中7 和20 号染色体各包括5 个区域,6、10、13、14、16 和20 号染色体均包括3个区域;在置信度0.99时,获得15个关联区域,分别位于9 条染色体(1、3、4、7、12、13、15、19 和20 号),其中7 和20 号染色体包括6 个区域,4号染色体包括2个区域。
2.3.3G-statistic 算法发掘遗传位点分析 采用G-statistic 方法分析与荚粒性状关联的位点,结果(图1C)表明,在置信度0.95 时,获得51 个关联区域,位于18条染色体(除5和18号外),其中7号染色体包括10 个区域,20 号染色体包括8 个区域;在置信度0.99时,获得16个关联区域,分别位于9条染色体(1、3、6、7、12、15、17、19 和20 号),其中7 号染色体包括6 个区域,3 和20 号染色体包括2个区域。
2.3.43 种算法共关联遗传位点分析 综合分析上述3 种算法获得的关联区域(表5),在置信度0.95 时,获得27 个一致性关联区域,位于15 条染色体(除5、8、10、11和18号外);在置信度0.99时,获得4 个一致性关联区域,分别位于4 条染色体(1、7、12 和19 号),其中15 个基因位于1 号染色体,57 个基因位于7 号染色体,82 个基因位于12号染色体,182个基因位于19号染色体。

表5 大豆荚粒性状不同算法一致性关联位点Table 5 Genetic loci of soybean pod and seed traits via different methods
2.4 BSA-seq与连锁定位一致性遗传位点发掘
综合分析本研究采用BSA-seq 方法获得的遗传位点以及课题组前期采用连锁定位方法(SoySNP6K 分析)获得的遗传位点[18]发现,在7 号染色体的10.45~11.59 Mb 和12.96~13.54 Mb 区段,19 号染色体的47.63~48.50 Mb 和49.61~49.91 Mb 区段以及20 号染色体的35.83~36.24 Mb 区段存在一致性。并且发现,20 号染色体SNP 标记ss715637629(物理位置为35 836 361),19 号染色体SNP 标记ss715635705(物理位置为47 636 564)、ss715635755(物理位置为48 040 350)、ss715635827( 物理位置为 48 375 196)、ss715635844( 物理位置为 48 501 612)、ss715635790( 物理位置为 48 196 242)、ss715635952(物理位置为49 618 939)和ss715635964(物理位置为49 699 850)在连锁定位和BSA-seq分析中同时被检测到,一方面证实了本研究结果的可靠性,另一方面为菜用和粒用大豆荚粒性状分子遗传改良提供了新的信息。
2.5 大豆荚粒性状候选基因筛选
为进一步筛选大豆荚粒性状候选基因,本研究在上述3 种算法0.99 置信水平共定位的染色体区段寻找到336 个荚粒性状候选基因的SNP 等位变异(表6),有40 个基因在DNA 水平存在外显子非同义突变,暗示其可能与大豆荚粒性状有关。

表6 BSA-seq一致性区间存在外显子非同义突变的候选基因Table 6 Candidate genes in the consistent regions with non-synonymous mutations in gene exons
在此基础上,利用供试RIL 群体亲本C813 和KN7的豆荚和籽粒不同发育时期转录组数据分析上述40 个基因,结果(图2、图3)发现,有15 个基因在豆荚和籽粒中表达量较高,其中包括Glyma.07G108700、Glyma.19G220000、Glyma.19G222200、Glyma.19G222300和Glyma.19G222400等;候选基因Glyma. 19G222200、Glyma. 19G222300和Glyma.19G224400随豆荚发育进程表达量呈现增加趋势。由此可见,上述15个候选基因在DNA水平存在外显子非同义突变,而且RNA 水平在豆荚中表达量较高,故认为其与大豆荚粒性状形成具有密切关系,尤其是随着豆荚发育进程表达量呈现增加趋势的候选基因Glyma.19G222200、Glyma.19G222300和Glyma.19G224400,可用于基因克隆以进一步证实其在大豆荚粒发育过程中的生物学功能。

图2 大豆荚粒性状候选基因在C813中的表达分析Fig. 2 Expressions of candidate genes related to soybean pod and seed traits in C813

图3 大豆荚粒性状候选基因在KN7中的表达分析Fig. 3 Expressions of candidate genes for soybean pod and seed traits in KN7
3 讨论
3.1 大豆荚粒性状一致性遗传位点的发掘与利用
BSA策略可用于存在遗传差异的多种群体,且可与传统定位或关联方法结合使用[21]。郭微[22]利用BSA-seq结合连锁定位方法将水稻耐盐碱QTLs缩小至2 号染色体的2 个区段;李君[23]利用BSAseq结合连锁定位方法寻找玉米圆斑病遗传位点;Vogel等[24]结合BSA-seq与连锁定位方法获得南瓜疫霉根腐病和冠腐病QTLs。本研究利用BSA-seq对大豆RIL 群体极端家系构建的2 个混池进行分析,经综合3 种算法关联分析,在0.95 置信水平将控制荚粒性状遗传位点定位在15 条染色体;同时,本课题组前期曾利用该RIL 群体,通过连锁定位方法将控制荚粒性状遗传位点定位在7 条染色体,表型贡献率5.28%~17.30%[18]。进一步比较BSA-seq 结果与连锁定位结果发现,二者在7、19和20 号染色体存在共定位区间,且19 和20 号染色体的多个SNP 标记被2 种策略同时检测到,充分证实了本研究结果可靠性和真实性。
关于20 号染色体控制大豆荚粒性状遗传位点,Hina 等[9]曾在该染色体的36.18~38.30 Mb 和35.92~38.14 Mb 区段检测到控制粒长与长宽比QTLs;Zhao 等[25]曾在该染色体35.23 Mb 附近检测到控制粒长QTLs;本课题组前期曾在该染色体35.84~36.23 Mb 区段检测到多环境、多性状共定位一因多效QTL(ss715637668~ss715637629),可同时控制大豆荚重、粒重、粒长和籽粒面积,解释8.92%~17.30%表型变异,而且在相应性状BLUP值的连锁定位中同时也被检测到,解释12.07%~16.81%表型变异[18]。本研究通过BSA-seq 技术,将控制大豆荚粒性状遗传位点定位到20 号染色体的34.99~38.10 Mb 区段,与上述连锁定位存在一致性区间。并且,为进一步利用上述一致性SNP标记,本课题组将该标记转换为PCR标记,经扩增供试RIL 群体亲本C813 和KN7,结果发现,能够按照预先设计扩增出目的条带(C813 可扩增500 bp 左右目的条带,而KN7 中没有该目的条带),继续扩增RIL 群体2 个混池(LP 和SP)的40个家系,发现LP 混池中有15 个家系可扩增出目的条带,而SP 混池中仅有4 个家系可扩增出目的条带,进一步扩增RIL群体其他家系将其划分为2种基因型,且与预期结果基本一致,既证实了本研究结果的真实性,也为大豆荚粒性状分子遗传改良提供了技术支撑。
3.2 控制大豆荚粒性状候选基因分析
关于大豆荚粒性状候选基因,目前研究报道较少。Wang 等[1]获得了可同时影响籽粒大小、脂肪和蛋白质含量的功能基因GmSWEET10a和GmSWEET10b,超表达2 个基因均可显著增加百粒重和脂肪含量;而沉默基因后则可显著降低百粒重和脂肪含量。最近,王明晓等[26]将控制豆科植物种子大小的基因划分为3 类,即转录因子类(如WRKY、AP2/ERF 和TIFY 家族等)、植物激素类(生长素、油菜素内酯、赤霉素和细胞分裂素等)和其他因子类(细胞色素P450 家族、泛素介导的蛋白水解、细胞壁转化酶抑制因子、肌醇单磷酸酶、ABC 转运蛋白等),但同时也指出,豆科植物种子大小非常复杂,目前有关其控制基因及其调控网络仍很欠缺。鉴于此,本研究在前述多年、多点鉴定RIL 群体6 个荚粒性状,并分别通过BSAseq 和连锁定位方法发掘遗传位点的基础上,进一步寻找控制荚粒性状候选基因,结果发现,在一致性关联区间内有336 个基因,并有40 个基因的外显子存在非同义突变,其中包括转录因子基因Glyma. 19G221700、Glyma. 19G222200、Glyma.19G224700和Glyma.19G236900以及其他因子类基因Glyma.19G219500、Glyma.19G224400和Glyma.19G226000等。
在上述候选基因中,Glyma.19G222200编码蛋白属于MYB 转录因子,该基因在供试大豆RIL群体亲本和混池间存在2 个非同义突变,且基因表达量随大豆荚粒发育而呈现上升趋势。拟南芥MYB 转录因子基因KUODA1参与植物叶片细胞的扩张,该基因突变后,可使转基因植株的叶片显著变小,而恢复植株的叶片与野生型基本相同,且基因超表达植株的叶片面积显著增大[27];拟南芥MYB 转录因子基因DRMY1参与植物细胞的扩张过程,该基因突变可引起一系列参与植物细胞壁生物合成与重塑的基因表达量发生改变,并使转基因植株的角果显著变短[28]。因此,本研究推测MYB 转录因子基因Glyma.19G222200可能通过影响其下游一系列与大豆荚粒生长发育相关基因的表达来控制荚粒发育,因而具有重要研究价值。
另外,植物体内的硝酸盐转运蛋白不仅具有运输亚硝酸盐的重要功能,而且可运输生长素、赤霉素、脱落酸和茉莉酸等多种物质,因而对植物正常生长发育至关重要。魏一鸣[29]通过研究玉米EMS 突变体发现,硝酸盐转运蛋白基因ZmNPF7.9发生单碱基的非同义突变(G/A 突变),可导致玉米籽粒变小,顶部凹陷且粒重下降;进一步分析发现,该突变体植株参与糖酵解、碳代谢及氨基酸合成的关键基因表达量降低,推测其通过影响籽粒中的营养物质运输和代谢进而导致玉米籽粒变小、粒重下降。在本研究中,候选基因Glyma.19G224400编码蛋白属于硝酸盐转运蛋白家族,该基因在供试大豆RIL 群体的2 个亲本间以及混池间存在3 个非同义突变,且基因表达量随大豆荚粒发育进程呈现上升趋势,因而推测其可能通过参与大豆荚粒中营养物质以及激素类物质运输进而影响荚粒发育。综上,本研究认为,上述候选基因在DNA 水平存在外显子SNP 非同义突变,而且随大豆荚粒生长发育呈现上调表达,说明其与荚粒生长发育密切相关,为进一步研究基因生物学功能奠定了重要基础。
