COL4A1基因相关脑穿通畸形1例报告并文献复习
2023-08-04蒋琼朱会王齐艳罗泽民龙萍萍曾兰石镜懿周家吉朱书瑶
蒋琼 朱会 王齐艳 罗泽民 龙萍萍 曾兰 石镜懿 周家吉 朱书瑶
脑穿通畸形是一种罕见的先天性中枢神经系统疾病,影像学上表现为脑实质缺损并与脑室相通。分为两型,1型称为假性脑穿通畸形(OMIM:175780),认为可能与妊娠晚期脑室周围出血或中枢神经系统感染等有关,是获得性疾病。2型称为真性脑穿通畸形,认为可能与妊娠中期神经元迁移异常有关,是先天性疾病[1]。脑穿通畸形常在婴儿期被诊断,存活者通常有严重的发育迟缓,癫疒间,脑积水等症状。COL4A1基因与COL4A2基因变异可导致脑出血及脑穿通畸形[2],COL4A1基因突变更为常见且症状更重。目前国外已报道200余例COL4A1基因相关脑病,国内报道少,仅10例[3-5],伴有脑穿通畸形仅2例[2]。COL4A1基因变异异质性较高,可导致视网膜动脉迂曲(OMIM:180000),遗传性血管病、肾病、动脉瘤和肌肉痉挛综合征(OMIM:611773),脑白质病变(OMIM:618564),伴有或不伴有眼部异常的脑小血管疾病(OMIM:17580),脑小血管病变伴脑出血(OMIM:614519)[6]。国内尚无表现型,影像学及基因型总结。本文报道2021年1月四川省妇幼保健院儿科确诊的1例COL4A1基因变异相关脑穿通畸形患儿的临床资料,并复习该病相关文献,分析并总结国内该基因变异相关脑穿通畸形的表现型,影像学及基因型特点。
病历资料
先征者,男,2岁9月,因“反复眼睑眨动2周”于2020年10月首次就诊于四川省妇幼保健院小儿神经内科门诊。2周前患儿无明显诱因出现眨眼,表现为2~6次/串,数十串/天,发作时伴或不伴眼球、头及躯干向右侧偏转,发作前进行的动作有终止,持续约2~10秒后自行停止,无面色青紫、四肢强直及抖动,发作过程中手中握持物体无坠落,有无意识障碍不详,发作后无困倦样表现。
查体:体重11 kg、身长75 cm、头围45.5 cm,外貌无特殊,四肢可见对称抬离床面,左侧肢体肌张力增高,右侧肢体肌张力正常。左侧肢体活动受限,使用频率较右手明显降低,喜右手握持物体及玩玩具,双侧病理征阴性,外生殖器及脊柱无畸形。G1P1,40+1周顺产娩出,无窒息病史。
出生头围:33 cm,体重3.2 kg,身长48 cm,自幼全面发育落后。抬头、独坐时间不详,1岁能扶站,能发音,几乎无眼神交流。父母体健,无亲缘关系,怀孕后期母亲院外产检发现胎儿双侧脑室不等大,未行产前诊断咨询。家族中无惊厥史,无同类病史记载。
既往史:新生儿期因黄疸住院治疗,好转出院。入院后实验室检查结果:血常规、肾功、心肌酶、血气分析、电解质、血糖、血氨、铜蓝蛋白、血脂、甲状腺功能、眼底检查、听力筛查均正常。腹腔、心脏超声、胸片、心电图未见异常。遗传代谢病血串联质谱及尿液有机酸气相色谱-质谱检测正常,染色体核型分析46,XY。
儿童神经心理发育评估报告单:发育商低下。磁共振平扫颅脑检查所见:1、颅内右侧大脑半球局部脑组织缺失,同侧白质纤维束明显较对侧减少。2、右侧枕叶体积较左侧枕叶小,以枕叶脑白质体积减小为主(图1)。
脑电图:幼儿期异常脑电图,基本节律变慢,节律差,双侧弥漫性不规则中高波幅混合漫波,间有多灶性棘波/尖波,右枕明显,双侧基本对称;监测中见双侧多导联散在尖波/棘波,多次阵发性广泛导联中波幅0.5~5 HZ棘慢复合波,多棘慢复合波连续发放,持续1~3秒,监测中未见明显发作(图2)。
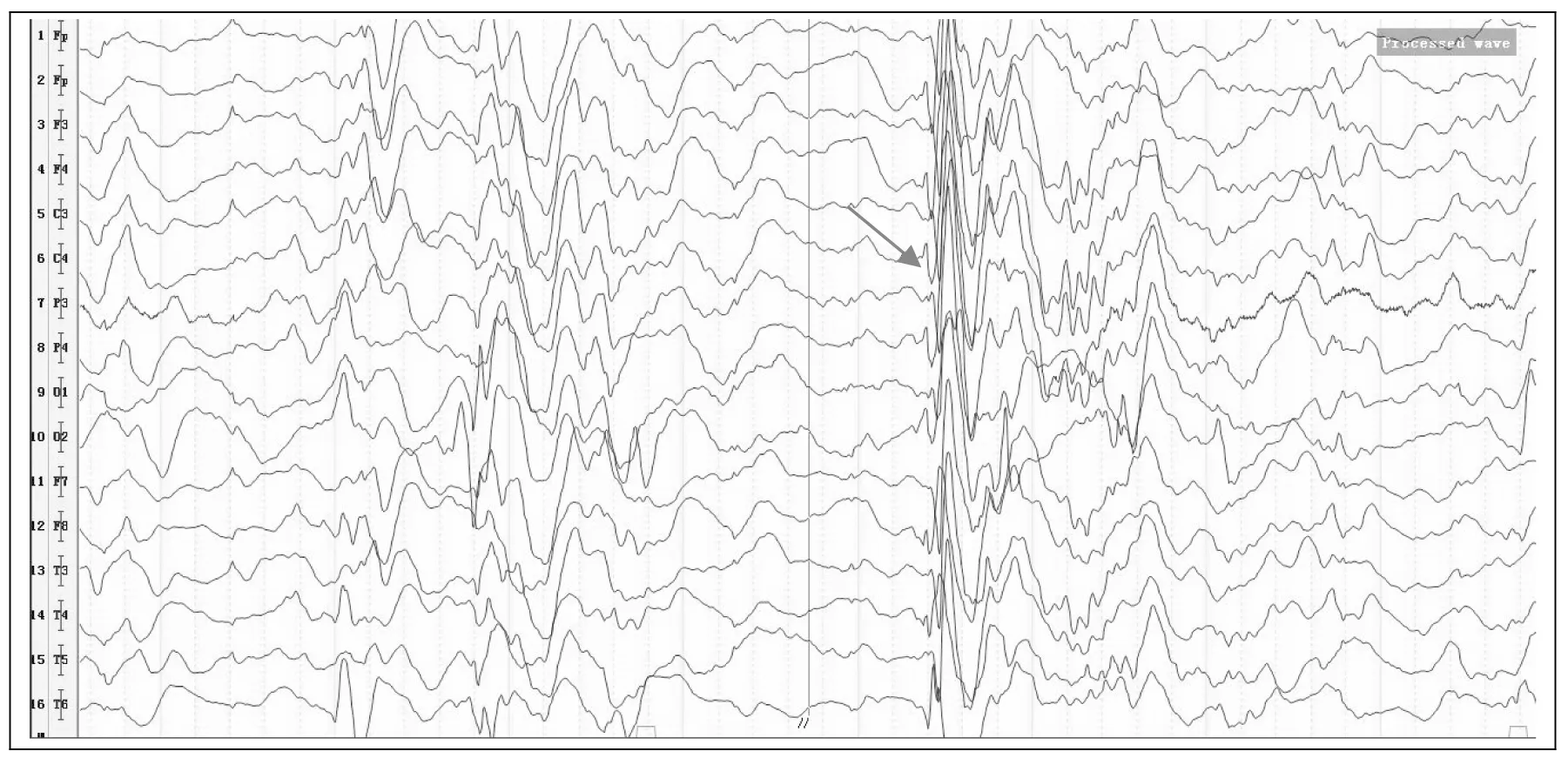
箭头显示双侧多导联散在尖波/棘波,多次阵发性广泛导联中波幅0.5~5 HZ棘慢复合波,多棘慢复合波连续发放
获得患儿父母知情同意后,该家系行家系Trio全外显子组基因测序,检测出先证者临床表型高度相关的基因COL4A1(NM_001845):c.2245G>A(p.G749S),杂合突变。Sanger测序验证结果显示其父母为野生型,无先证者相关临床表型,根据美国医学遗传学和基因组学学会变异分类(ACMG)指南[7]:c.2245G>A为经双亲验证的新发变异,符合PS2。已有文献报道[7-8]。在2名脑穿通畸形患者中检测到该变异(PMID:11043343),符合PS4。该变异在ExAC、gnomAD、千人基因组亚洲人群数据库中没有收录,符合PM2。PolyPhen-2(评分=1)、SIFT、Provean及Mutation Taster等蛋白危害性预测软件提示该变异为有害,符合PP3。该变异评估为致病突变(PS2+ PS4+PM2 +PP3)。结合患儿的临床表现和基因分析结果,此患儿确诊为脑穿通畸形1型。
入院后给与丙戊酸钠口服液治疗后,仍有反复癫疒间发作出院。门诊增加抗癫疒间药物托吡酯、左乙拉西坦,随访1年9月,癫疒间发作稍减少,约2~4次/月,仍有左侧肢体活动受限,不能扶走。2岁时会无意识发“mama”音。有不恰当社交方式(与人打招呼表现为拍人),不听指令,无眼神及语言交流,抗拒陌生人及环境,对同龄人不感兴趣。CARS:36分(重度异常),建议教育训练,家属拒绝,仅愿意继续抗癫疒间治疗。
讨论
脑穿通畸形是一种先天性脑疾病,特征是孔洞形成或没有灰质包裹充满脑脊液的囊肿形成。生后临床表现癫疒间、瘫痪、智力障碍[6]。1型常单侧发生,由产伤、感染、出血等原因引起,2型考虑为先天性疾病,常对称发生。Berg等[1]在1983年首次报道了累及3人的家系脑穿通畸形,推测染色体显性遗传可导致遗传性脑穿通畸形。Gould等[8]2005年通过杂合子突变体小鼠发现COL4A1基因与COL4A2基因变异可导致小鼠脑出血及脑穿通畸形,并报道两例COL4A1基因变异所致脑穿通畸形的患者,其中1例与本研究一致。Gasparini等[9]2017年在该基因的相同位点报道1例脑穿通畸形患者。研究发现,在218个脑畸形病人中发现56人携带COL4A1基因变异,其中29人为脑穿通畸形,证明了COL4A1基因功能变异在儿童脑穿通畸形起重要作用[6]。
IV型胶原蛋白几乎是所有基底膜的主要成分。研究发现[10],COL4A1基因编码IV型胶原蛋白的 alpha-1(a1)亚基和COL4A2基因编码a2亚基;其中人类COL4A1基因位于13q34,高度保守基因;由48个外显子组成,编码含1 669个氨基酸;a1a1a2形成异质三聚体IV型胶原蛋白;a1链有三个功能域,分别为氨基端(7S)结构域、三螺旋结构域(THR)及羧基端(NC1)结构域;其中由甘氨酸(Gly)-Xaa-Yaa重复形成的三螺旋占a1链结构的75.9%[11-12]。Gly为无侧链氨基酸,在三螺旋结构中起重要的稳定重要,突变可导致链间氢键被破坏,造成三螺旋稳定降低[13],可引起突变小鼠基底膜出血。COL4A1基因变异胎儿尸检发现颅内小血管微出血及微血管血栓形成,可能是引起脑畸形的病理机制[14]。研究显示,基因变异小鼠通过改变分娩方式,可减少颅内出血,但人类尚无相关证据[8]。
查阅文献发现,家族性发病可累及8人之多[15]。症状由仅视野缺损到脑穿通畸形、偏瘫及癫疒间,其中3人后经基因检测确认为COL4A1基因p.G749S突变(与本研究一致)。显示出COL4A1基因突变在家族间及家族内存在异质性及外显不全。国内COL4A1基因变异相关神经系统受损除脑穿通畸形外脑白质病变,神经系统以外以血管病、肾病常见,未见孤独症相关表现。但需注意除COL4A1/COL4A2基因突变与脑穿通畸形有关,还包括COLGALT1,TUBA1A基因突变已有脑穿通畸形报道[16-17]。
本病无特效治疗,主要为对症治疗。其中癫疒间主要发作形式以局灶性发作为主,偶有痉挛性发作报道。针对药物难治性癫疒间,功能性大脑半球切除术手术可能是一个有效选择[18],但相关病例较少。
本例患儿产前检查有脑室不等大,生后有癫疒间、偏瘫、全面发育落后,合并自闭症表现,MRI示脑穿通畸形,结合Trio-WES检查COL4A1基因存在c.2245G>A(p.G749S)突变,诊断COL4A1基因相关脑穿通畸形明确。胎儿期有脑室增宽,出生后有癫疒间,偏瘫,全面发育落后,需考虑COL4A1基因变异可能,及时完成基因测序以提供有效的遗传咨询。
