Mechanism of nickel-catalyzed hydroalkylation of branched 1,3-dienes
2023-07-18MingqiangLiuDeguangLiuZheyuanXuHaizhuYuandYaoFu
Mingqiang Liu ,Deguang Liu ,Zheyuan Xu ,Haizhu Yu ,and Yao Fu
1Hefei National Research Center for Physical Sciences at the Microscale, CAS Key Laboratory of Urban Pollutant Conversion, Anhui Province Key Laboratory of Biomass Clean Energy, iChEM, University of Science and Technology of China, Hefei 230026, China;
2Department of Chemistry, Center for Atomic Engineering of Advanced Materials, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei 230601, China
Abstract:With the development of algorithms and theoretical chemistry,quantum chemical calculations have been used to explain and predict various chemical experiments.The hydroalkylation of conjugated olefins catalyzed by nickel is an important type of organic chemical reaction,and its mechanism has always been the focus of organic chemists.In this paper,a hydroalkylation reaction developed by the Mazet research group was studied in detail by means of density functional theory (DFT),and a possible mechanism model of the reaction was obtained.In this context,the attractive regioselectivity of the reaction was explored and rationally explained.
Keywords:DFT;hydroalkylation;1,3-dienes;nickel catalysis;mechanism
1 Introduction
C-C bond formation has always been an enduring topic in organic chemistry[1,2].Among various types of C-C bond formation reactions,the hydroalkylation of alkenes has attracted much attention due to its advantages,such as high atom economy and mild reaction conditions[3-6].Compared to normal alkenes,the hydroalkylation of dienes might occur at different sites,resulting in a more diverse reaction selectivity.Therefore,the control of its selectivity has always been a matter of organic chemistry.Since 2004,Hartwig et al.[7]has pioneered the palladium-catalyzed 1,4-hydroalkylation of dienes (Fig.1a).In the past two decades,many excellent works (such as expanding the range of substrate alkenes and nucleophiles,other catalysts and bimetallic catalysis,control of regioselectivity,etc.) have made great contributions to hydroalkylation reactions[8-20].For example,Zhou et al.[21]introduced an unstable nucleophile to complete the hydroalkylation under nickel catalysis;Malcolmson et al.[22-24]extended the range of diene substrates to branched dienes and internal dienes;and Zi et al.[25]developed a copper/palladium dualcatalyst system to enable the reaction to achieve high enantioselectivity (Fig.1b-d).In 2020,the Mazet group[26]reported two complementary regiodivergent hydroalkylations of conjugated dienes with Ni catalysts by modifying the type of alkylation substrate (amide and imide).That is,when amides are used as nucleophiles,1,4-hydroalkylation is favored,while when imides are used,3,4-hydroalkylation mainly occurs (Fig.1e).
According to the reported literature[27-34],the hydroalkylation of 1,3-dienes generally consists of two processes,hydrogenation and alkylation.Ligand exchange of catalyst ligands by dienes is a common and easily occurring process[35,36].After the formation of intermediateA(Fig.2),Ni-H speciesBcan possibly be formed by the oxidative addition of methanol toA(proton transfer can also occur to directly form the M-H species[23,37-41]) and then produce metal-π-allyl complexDvia Ni-H migratory insertion with the dienes (Fig.2,path I).Furthermore,complexDcould also be formed by the concerted protonation oxidation addition step of intermediateAvia transitional stateC(Fig.2,path II)[42-44].After hydrogenation,there are two possible pathways for C-C bond formation.One is to form a Ni-C bond with nucleophiles to attack the metal center and then undergo a reduction elimination step to form productG(Fig.2,path III)[45,46].The other involves the nucleophilic attack of the diene with carbonation from the outside direction (E→G,path IV)[37,38,43,44].Finally,ligand exchange betweenGand the diene substrate occurs,thus realizing the catalytic cycle.

Fig.2.Possible reaction mechanism of the Ni-catalyzed hydroalkylation of 2-phenyl-1,3-diene.
2 Materials and methods
All calculations were performed with Gaussian 16,Rev.C.01[47].The B3LYP functional[48-51]with the D3 version of Grimme’s empirical dispersion with Becke-Johnson damping(GD3BJ)[52,53]was used for geometry optimization of all structures.The def2-SVPP[51]basis set was employed on all elements.Frequency analysis was performed at the same level to confirm that the optimized structures are local minima (have no imaginary frequency) or transition states (have only one imaginary frequency) and acquire the thermal correction to Gibbs free energy.Single-point energy calculations were conducted with the M06 functional[51,54,55]and the def2-TZVP[56,57]basis set.The solvent effects were considered in all calculations with the SMD[58]solvation model (with tetrahydrofuran solvent).All transition states were further confirmed by intrinsic reaction coordinate (IRC)[59,60]calculations to ensure that the correct reactants and products were connected.The relative Gibbs free energy in calculations was corrected according to recent studies[61-64].Herein,conversion of the computed free energy at the 1 atm standard state (ΔG°atm) to that at the 1 mol/L solution state (ΔG°M) follows the equation ΔG°M=ΔG°atm+ΔnR1Tln(R2T),whereinR1=8.314 J·K-1·mol-1,and Δndenotes the molar change of the primitive step.Meanwhile,artificially increased pressure (P=(d/M) ×R2T) proposed by Martin et al.was used to reduce the overestimation of translational entropy[61],in whichMandddenote the solvent molar mass and density,respectively,andR2=0.082 L·atm·K-1·mol-1.For the model reaction in Fig.1e[26],T=298.15 K,and for the THF solvent,M=72.11 g·mol-1,d=786 g·L-1.To this end,ΔG°corr=ΔG°M+ΔnR1TlnP=ΔG°M+Δn× 3.3=ΔG°atm+Δn× 5.2.Unless otherwise stated,all energies are corrected Gibbs free energy in kcal/mol.The relevant optimized structures were drawn using CYLview software[65].
The studied model experiment.In accordance with Mazet’s experiments[26],the Ni(cod)2/L1-catalyzed hydroalkylation of 2-phenyl-1,3-diene with amides or imides in tetrahydrofuran (THF) was used as the modeling reaction in the theoretical calculations (Fig.3).

Fig.3.Model reaction in the theoretical calculations.
3 Results and discussion
3.1 The hydroalkylation mechanism of the amide system
The reaction mechanism of the amide is first studied (the 1,4-addition process is favored).Regarding the initial state of the nickel catalyst,Ni(cod)2(cod=cycloocta-1,5-diene) (IN1)could undergo ligand exchange or coordination with the phosphinooxazoline ligand,solvents,dienes and potential additives.The results (Fig.S1) demonstrate that Ni-diene (IN2)formed via ligand exchange with the substrate is more feasible than all other cases.This result is in line with chemical common sense,and thus,IN2is set as the energy reference.
Given the plausibility of a prior hydrogenation step in the hydroalkylation reactions[21,23,37-41,66-70],we examined the energy demands for the formation of Ni-H species via the oxidative addition of methanol onIN2.Therefore,the energy calculation of the acid-base reaction between the amide and the potassium methoxide was carried out,and the generation of methanol and the zwitterionicK-amidewas exergonic by-1.4 kcal/mol (Fig.4a).Next,the corresponding transition state of the oxidative addition of methanol toIN2needs to overcome a high energy barrier of 42.3 kcal/mol (Fig.4b,TS1-c).In addition to the direct oxidative addition step[21,66-70],it is possible that proton transfer occurs directly from coordinated methanol to the alkene group (Fig.4b,TS1-a).However,the energy barrier is still unreachable in the reaction environment (ΔGTS1-a=29.8 kcal/mol).Considering that the transition state is mainly related to the proton transfer of methanol,the binding of a Lewis base (i.e.,K-amide) to oxygen is anticipated to be helpful[71-73].The structure optimization of the related transition state indicates a relatively lower energy barrier of 24.6 kcal/mol (Fig.4b,TS1-b).Herein,it is interesting to note that the coordination of oxygen of methanol is an endergonic step (Fig.S4,IN-inner,predominantly due to the significantly increased steric hindrance),so the possibility of an external hydrogen migration is considered (Fig.4b,TS1)[42-44].Satisfyingly,the energy barrier is further reduced to 19.6 kcal/mol.For comparison,the hydrogenation of the C1-site of the substrate is relatively more difficult,with an energy barrier of 22.5 kcal/mol (Fig.S5,TS1-h1).

Fig.4.The Gibbs free energy profiles of the hydrogenation step start from IN1.
Subsequently,the nickel-π-allyl speciesIN3was formed after the hydrogenation step.Herein,IN3could be viewed as a tightly bound ion pair,with a positively charged nickel-allyl moiety and a negatively charged methoxy-potassium amide moiety (structureIN3in Fig.5).FromIN3,different pathways might be responsible for the alkylation step on the C1/C3-site,depending on whether the anionic MeO-andKamideis released or not.As shown in Fig.5,removing the anionic MeO-andK-amidegenerates a slightly more stable intermediateIN4,while the coordination of the oxygen atom of anionic MeO-andK-amideto the nickel center generates a significantly more stable intermediateIN4-a.FromIN4,the relative Gibbs free energy of the C-C bond formation transition state on the C1-and C3-site is 25.7 and 30.6 kcal/mol,respectively (Fig.5).In contrast,the C-C bond formation fromIN4-aresults in a significantly higher energy barrier,and the Gibbs free energy of the related transition state reaches 37.7 kcal/mol.The high energy barrier of this process is presumed to originate from the high steric hindrance around the nickel center.For the same reason,the direct C-C bond formation from the carbonation coordinated intermediate (IN4-b→TS2-A-c,Fig.5) via a reductive elimination mode is remarkably more energy-demanding.
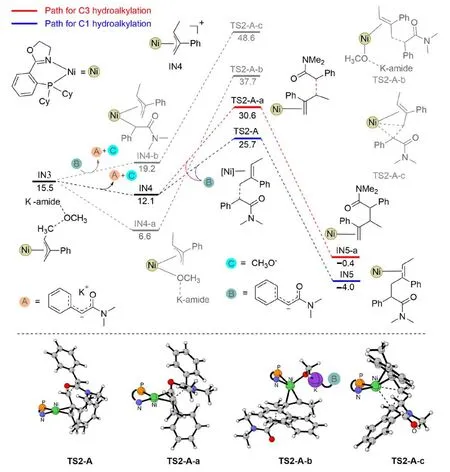
Fig.5.The Gibbs free energy profiles for the C-C bond formation steps from IN3.
According to the aforementioned results,in the amide system,the hydrogenation of the C4-site (over the C1-site) of the diene substrate occurs favorably in an outer-sphere mode and in the presence of the Lewis-baseK-amidemediation.The energy barrier is 19.6 kcal/mol.Next,the formed hydrogenation product dissociates the anionic MeO-andK-amidemoiety,with the formation of cationic intermediateIN4.After that,C-C bond formation occurs favorably on the C1-site(over the C3-site),with an energy barrier of 13.6 kcal/mol.The final catalyst regeneration step occurs,releasing the 1,4-hydroalkylation product as a result.For clarity,an overview of the transformation is shown in Fig.6.According to this mechanism,both the rate and regioselectivity determining steps lie in the C-C bond formation step.

Fig.6.The Gibbs free energy profile of the most feasible hydroalkylation of the 2-phenyl-1,3-diene with the amide reagent.
3.2 The hydroalkylation mechanism of the imide system
According to Mazet’s experiments[26],different regioselectivity occurs in the presence of amide or imide substrates,and therefore,we calculated the detailed energy profiles of the imide system (3,4-addition protocol was developed) (Fig.7).For clarity,the full calculation details of the hydrogenation and alkylation steps are given in Fig.S7 and Fig.S8,respectively.The overall transformation of the imide system is similar to that of the amide system.Due to the exothermicity of the acidbase interaction of KOMe and the imide substrate,the formation ofK-imideis a thermodynamically feasible process (Fig.S3).In this context,the photon transfer of methanol to C4 of the diene substrate in the imide system highly resembles that of the amide system,except Kimide functions as a Lewis base.After that,the dissociation of the anionic MeO-andKimideleads to the cationic intermediateIN4.In the subsequent C-C bond formation step,C3-alkylation is more favorable than C1-alkylation (see Fig.S8),which is consistent with the experimental selectivity[26].It is worth noting that due to the low energy barrier of the C-C bond formation steps in the imide system,the rate-determining step is the first hydrogenation step (with an energy barrier of 19.3 kcal/mol),while the regioselectivity determining step is the subsequent C-C bond formation step.

Fig.7.Energy profile of the main transformations in the most feasible hydroalkylation pathway of the imide system.
3.3 Discussions on the regioselectivity of the hydroalkylation
According to the aforementioned results,the regioselectivity could possibly be controlled in both hydrogenation (at the C1-or C4-site) and alkylation (at the C3-or C1-site) steps.Herein,the precursors of the C1-and C4-hydrogenation steps areIN2andIN2-a,respectively (Fig.8).The relative energy ofIN2is 0.5 kcal/mol higher than that ofIN2-a,while the electron density of the C4 atom inIN2(precursor ofTS1,-0.514) (Fig.8,C4) is relatively higher than that of the C1 atom inIN2-a(precursor ofTS1-h1,-0.466) (Fig.8,C1).Both the relatively higher concentration ofIN2and the relatively higher nucleophilicity of the C4 atom contribute to preferential C4 hydrogenation (19.6 kcal/mol vs 22.5 kcal/mol).

Fig.8.The Lewis structure,relative Gibbs free energy (ΔG° in kcal/mol),and optimized geometry of IN2 and IN2-a.
After C4 hydrogenation,the C1-or C3-alkylation step might generate 1,4-or 3,4-hydroalkylation products,respectively.As shown in Fig.9a,the nucleophilic attack of the amide anion on the C1-site results in an eclipsed conformation to ensure that the two relatively bulky groups (i.e.,phenyl and amide groups) keep away from the sterically hindered ligands on the Ni center (structure Ⅰ).In contrast,the C3-atom bears a methyl and a benzyl group.Great steric hindrance is present on the reaction center,and in this scenario,the staggered configuration is relatively more plausible(structure Ⅱ vs Ⅰ in Fig.9a).Due to the relatively higher steric hindrance,C3-alkylation is less favored than the C1-alkylation pathway,and thus,the 1,4-hydroalkylation product dominates in the amide system.The results correlate with the experimental outcome[26].

Fig.9.(a,b) Illustrative diagram for the configuration of the C1-and C3-alkylation transition states.The nickel on the other side is omitted for ease of observation.(c) The relative Gibbs free energy (ΔG° in kcal/mol),the NBO charge of the carbonyl group and the optimized geometry of Ⅰ,Ⅱ,Ⅲ,Ⅳand Ⅴ.
Compared to the amide systems,the C1-and C3-alkylation transition states in the imide systems adopt a similar eclipsed and staggered configuration (structure Ⅲ and Ⅳ in Fig.9b).Nevertheless,distinct from the sterically controlled configuration of the C3-alkylation with amide (structure Ⅱ in Fig.9a),the staggered configuration of the C3-alkylation with imide results in a sterically hindered mode.As shown in the structure in Fig.8,the imide group in Ⅳ lies trans to the hydrogen on the C3 atom of the substrate,which is anticipated to induce significantly higher steric hindrance in the reaction center.However,the relative energy of structure Ⅴ is lower than that of the sterically more feasible structure Ⅳ (analog of Ⅱin the amide system,Fig.9).The results indicate that extrastability induced such an uncommon configuration.Indeed,the optimized geometry (Fig.9c) of the C3-alkylation precursor of the imide system shows a π-π stacking interaction between the C=O bond of the imide group and the phenyl group of the allylic substrate (Fig.S10,and the distance between the groups is~3.3 Å).The electron-deficient phenyl group (due to the π-π conjugation and the positive charge on the allylic moiety) could be stabilized by the electron-rich carbonyl groups.Indeed,the NBO charges of the carbonyl groups in Ⅲand Ⅴ are 0.273 and 0.284,respectively (Fig.9c),verifying the electron transfer from the carbonyl groups to the allylic groups.Therefore,in the imide system,the carbonyl group of the imide and the benzene ring of the branched diene form an orbital interaction during the alkylation process,resulting in preferential C3-alkylation.In other words,the electronic effect results in the predominance of the 3,4-hydroalkylation over the 1,4-hydroalkylation pathway in the imide system.The regioselectivity of this system correlates with the experimental outcome[26].
To further verify the above conclusions,we performed distortion/interaction analysis with the transition states of the selectivity-determining step,and the relevant results are presented in Fig.S11.As expected,in the amide system,the distortion promoted the dominance of 1,4-hydroalkylation.In the imide system,the interaction between the fragments made 3,4-hydroalkylation more favorable.
4 Conclusions
In conclusion,the mechanism of Ni-catalyzed hydroalkylation of Mazet with amide/imide was systematically explored with the aid of density functional theory calculations.After comparing the different pathways,both systems adopt the same reaction mechanism,consisting of four main steps:hydrogenation with an in situ formed Lewis base (K-amide/Kimide),heterogenous bond dissociation with the formation of a cationic allylic coordinated intermediate,nucleophilic attack with amide/imide,and the final catalyst regeneration steps.C4 hydrogenation is favored over C1 hydrogenation in both systems due to the relatively stable precursor and the higher electron density on the reaction site.In this context,C1-alkylation is favored in the amide system due to the steric benefits,while C3-alkylation is favored in the imide system due to the electronic advantages induced by π-π stacking between the carbonyl group and the phenyl group of the substrate.
Supporting information
The supporting information for this article can be found online at https://doi.org/10.52396/JUSTC-2023-0031.The supporting information includes nine figures and Cartesian coordinates of the calculated structures.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21732006,51821006,21927814),the CAS Collaborative Innovation Program of Hefei Science Center (2021HSC-CIP004),and the University Synergy Innovation Program of Anhui Province (GXXT-2021-023).The numerical calculations were performed on the supercomputing system in the Supercomputing Center of the University of Science and Technology of China.
Conflict of interest
The authors declare that they have no conflict of interest.
Biographies
Mingqiang Liuis a master’s student under the supervision of Prof.Yao Fu at the University of Science and Technology of China.His research mainly focuses on DFT calculations of the reaction mechanism.
Haizhu Yureceived her Ph.D.degree from the University of Science and Technology of China.She is currently a Professor at Anhui University.Her research focuses on reaction mechanism simulation,structureactivity relationships,and anticancer metal nanoclusters.
Yao Fureceived his Ph.D.degree from the University of Science and Technology of China in 2005.He is currently a Professor at the University of Science and Technology of China.His research focuses on physical organic chemistry,green organic synthesis,and the biomass chemical industry.
杂志排行

中国科学技术大学学报的其它文章
- Recent progress on diaCEST MRI for tumor imaging
- The mystery of Li2O2 formation pathways in aprotic Li-O2 batteries
- Magnetic modification of lanthanide-based upconversion nanocrystals for fingerprint information recognition
- Mercaptopropane-assisted synthesis of graphitic carbonsupported Pt nanoparticles for enhancing fuel cell start-stop performance
- Amorphous TiO2 ultrathin nanosheet for stable high-rate lithium storage
- Polymerized small molecular dyes providing nanoparticles with stable photosensitivity for eradicating cancer cells