超高效液相串联质谱法测定牛奶中8 种氟喹诺酮类兽药残留
2023-07-09张瑞婷
张瑞婷
(北京市产品质量监督检验研究院,北京 101300)
氟喹诺酮类药物为白色或淡黄色晶型粉末,属于广谱抗生素,常被用于预防和治疗奶牛疾病[1]。兽药的过量使用或不当使用与公众的健康息息相关,长期、低水平的接触方式不仅导致细菌产生耐药性,还会产生各种慢性、蓄积性毒害,给消费者带来潜在危险。因此,建立兽药残留的分析方法对于风险监控是十分必要的[2]。目前,兽药残留量的检测方法主要有金标检测法、酶联免疫吸附法、高效液相色谱法、气相色谱法和质谱法等[3]。超高效液相串联质谱法的优点是选择性强、应用范围广、分离效果好、灵敏度高和能自动化操作[4]。其可以把待测组分分离与结构的定量分析有机结合在一起,特别适用于药物残留量和药物的确证分析[5]。
1 材料与方法
1.1 仪器与试剂
超高效液相串联质谱仪(Waters 公司);氮吹浓缩仪(Organomatian Associates 公司);冷冻离心机(Sigma 公司);Oasis HLB 固相萃取柱(200 mg,6 mL,Waters 公司)。
8 种喹诺酮类兽药标品:恩诺沙星、诺氟沙星、环丙沙星、司帕沙星、氧氟沙星、奥比沙星、萘啶酸和噁喹酸,均购于德国DR 公司;乙腈、甲醇、正己烷和甲酸(HPLC 级);超纯水(Mili-Q 公司)。
1.2 溶液的配制
1.2.1 标准储备溶液
精密称量各种药品标准品10 mg(精确到0.1 mg)于10 mL 容量瓶(棕色)中,用乙腈超声溶解并定容至刻度(其中噁喹酸需加少量NaOH 溶液溶解),配制成1.0 mg·mL-1标准储备溶液,4 ℃冰箱避光保存。
1.2.2 混标工作溶液
取洗净烘干的50 mL 棕色容量瓶,加入500 μL储备液,乙腈定容至刻度,配制成浓度为10.0 μg·mL-1的混标中间溶液,置于4 ℃冰箱冷藏避光保存。
1.2.3 流动相
0.3 %甲酸水溶液:准确吸取3 mL甲酸至1 000 mL容量瓶,用超纯水定容至刻度,现配现用。流动相:乙腈∶0.3%甲酸=10 ∶90。
1.3 样品前处理
1.3.1 提取
将牛奶样品轻晃混匀后称取5.00 g(精确到0.01 g),装入50 mL 的PP 管中,依次加入无水硫酸钠5 g,1%的甲酸-乙腈溶液20 mL,轻柔涡旋混匀,充分反应。超声波提取时,保持处于冰浴环境中,离心转速5 000 r·min-1,时间5 min,之后取上清液于另一洁净的离心管中。将剩余残渣重复一次上述操作,取上清液。吸取10 mL 提取到的上清液,加入20 mL 乙腈饱和的正己烷,振荡静置分层弃去正己烷层,氮气吹至近干。
1.3.2 净化
HLB 固相萃取柱(200 mg,6 mL)使用前用6 mL甲醇洗涤、6 mL 水净化,将提取的上清液以2 ~3 mL·min-1的速度过柱,过两次后弃去滤液,用2 mL 5%的甲醇溶液淋洗,弃去淋洗液,减压真空泵抽取5 min,在吸附柱中加入5 mL 甲醇洗脱液,5 000 r·min-1离心1 min,将收集到的洗脱液在氮气下吹至近干后,加入1 mL流动相溶液溶解,经0.22 μm微孔滤膜过滤,以备上机分析测定使用。
1.4 测定条件
1.4.1 色谱条件
色谱柱:Acquity UPLC C18色谱柱(2.1 mm×50 mm,1.7 µm BEH 颗粒,美国沃特世公司);流动相:A 相为乙腈,B 相为0.3% 甲酸水溶液;流速:0.3 mL·min-1;柱温:30 ℃;进样体积:5 μL。
1.4.2 质谱条件
离子源:电喷雾电离-正离子模式(ESI+);检测方式:MRM(多反应监测);毛细管电压:3.5 kV;脱溶剂气流量:800 L·h-1;锥孔气流量:120 L·h-1;碰撞气:氩气;碰撞气压:0.18 Pa。
2 结果与分析
2.1 提取剂的选择
在酸性提取溶剂中,氟喹诺酮类药物更容易被萃取;乙腈属于极性溶剂,可避免从样品中提取过多的脂肪,同时具有很好的蛋白沉积效果,可沉积样品中99%的蛋白,避免其他杂质的干扰。因此,初步确定采用乙腈为提取溶剂。
本实验对纯乙腈、1%乙酸-乙腈、1%甲酸-乙腈3 种提取剂提取效果进行了对比,结果显示酸化乙腈提取效果更佳。其中1%甲酸-乙腈、1%乙酸-乙腈的提取效果相差不大,但1%甲酸-乙腈提取后的色谱峰更加尖锐,故确定最终提取溶剂为1%甲酸-乙腈。
2.2 流动相的选择及质谱条件的优化
以不同比例的乙腈-水为流动相进行梯度洗脱时,为了提高正离子化效率并改善峰形,同时考虑到分析组分中大多含有氨基和羧基基团,所以分别尝试了在水相中加入0.1%甲酸、0.2%甲酸、0.3%甲酸、5 mmol·L-1甲酸铵、10 mmol·L-1甲酸铵进行洗脱。对比结果显示,流动相中加入0.3%甲酸时,样品由结合态变为游离态,灵敏度高,分离效果好。
质谱参数的优化是通过ESI 探针分别将8 种兽药(浓度1 mg·L-1)的混合标准品工作液在正电离模式下用质谱蠕动泵自动进样,对每一种兽药进行母离子全扫描确定各种待测兽药的分子离子,而后取其分子离子为母离子,对其进行全扫描。选取丰度最强、干扰最小的两个离子用于目标分子的定量和定性分析,表1 为8 种兽药质谱分析优化参数。
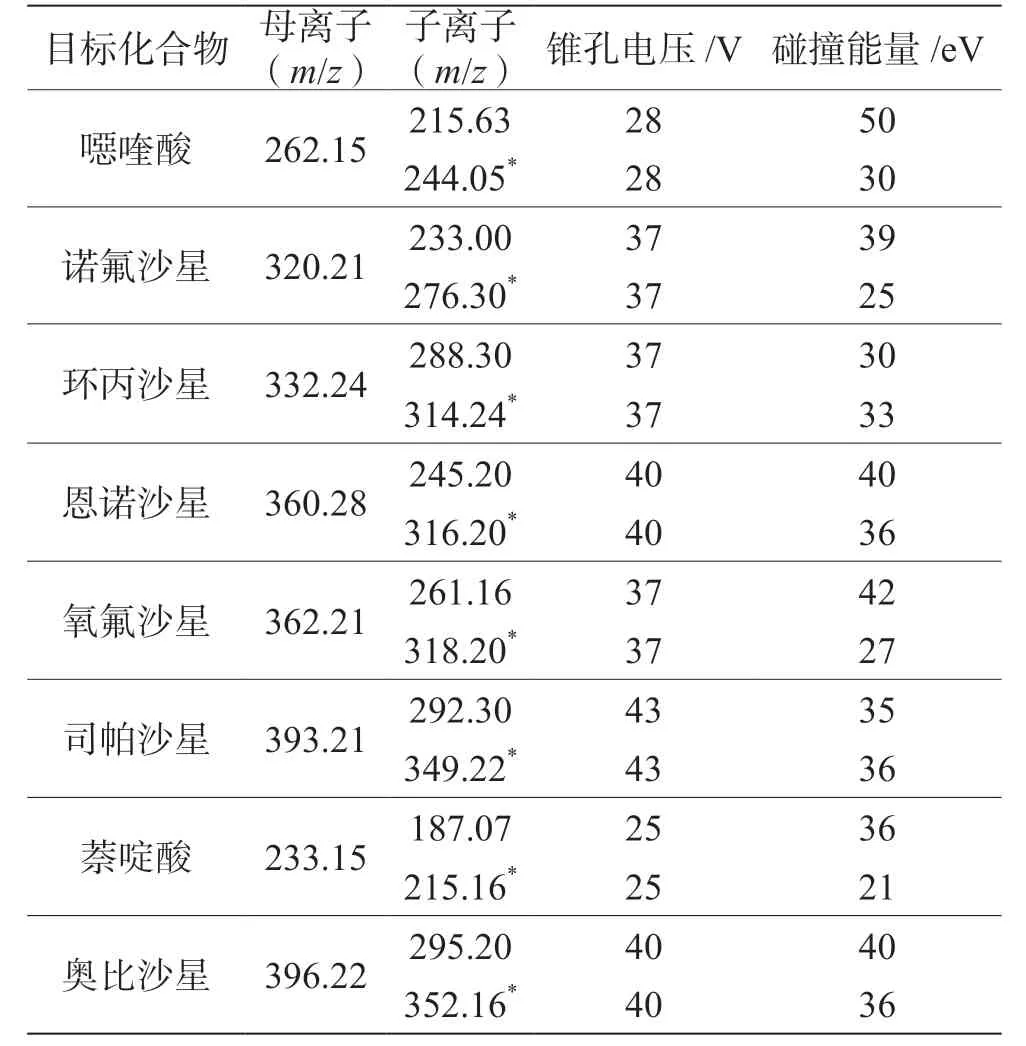
表1 8 种兽药质谱分析优化参数
2.3 线性范围及检出限
将8 种待测兽药配制成一系列标准工作溶液,绘制标准工作曲线,对目标物质进行定量分析。分别得到待测兽药的标准曲线回归方程、定量限及相关系数,结果表明在0.5 ~500.0 μg·mL-1,R2>0.996,8 种兽药的回归方程均线性良好。通过逐级稀释,根据信噪比(S/N)等于10 确定为定量下限,结果见表2。

表2 8 种兽药的线性方程、相关系数
2.4 回收率与精密度
在待测样品中添加8 种兽药的混标工作溶液,分别在10 μg·kg-1、50 μg·kg-1、100 μg·kg-13 个浓度水平下进行回收率实验,同时每个浓度水平下进行6次平行实验,计算精密度。结果见表3。结果显示,该方法的平均回收率为81.3%~98.8%,RSD 为2.3%~9.8%,表明本方法的回收率和RSD 均在标准范围内,符合多残留分析的要求。

表3 牛奶中8 种兽药的回收率与精密度
2.5 实际样品检测
在市场上随机购买15 份不同批次牛奶样品,经检测发现1 份阳性样品。恩诺沙星检出值12.1 µg·kg-1,其他药物无检出。该例阳性样品兽药残留量未超过GB 31650—2019 规定的最高残留限量,可认为是合格样品。
3 结论
本实验建立了一种简单、灵敏、快速的8 种兽药残留分析方法,通过对净化方法、流动相、质谱条件等一系列实验条件的探索优化,建立了UPLCMS/MS 同时检测牛奶中8 种氟喹诺酮类兽药的分析方法。该方法克服了检测种类少、检测时间长、实验成本高等问题,适用于牛奶样品多残留的快速筛查和检测。
