基于QuEChERS-超临界流体色谱-串联质谱法的茶叶中甲胺磷及乙酰甲胺磷对映体拆分及定量
2023-07-06王丽
王丽


摘要采用QuEChERS法提取,在以Chiral-Pak IC-3为色谱柱的超临界流体色谱上拆分,大气压化学电离后串联四极杆质谱法测定。结果表明,茶叶中(+)-甲胺磷、(+)-乙酰甲胺磷的检出限为5 μg/kg,定量限为10 μg/kg,平均回收率为70.53%~110.61%,相对标准偏差(RSD)为2.92%~13.30%。该方法能够有效拆分和定量甲胺磷和乙酰甲胺磷对映异构体。
关键词QuEChERS;超临界流体色谱-串联质谱;茶叶;甲胺磷;乙酰甲胺磷;对映异构体
中图分类号TS272文献标识码A
文章编号0517-6611(2023)11-0156-07
doi:10.3969/j.issn.0517-6611.2023.11.038开放科学(资源服务)标识码(OSID):
Separation and Quantification of Methamidophos and Acephate Enantiomer in Tea Based on QuEChERS-Supercritical Fluid Chromatography-Tandem Mass Spectrometry
WANG Li(Beijing Institute of Food Inspection and Research (Beijing Food Safety Monitoring and Risk Assessment Center),Beijing 100094)
AbstractExtraction was performed by QuEChERS method,separated on supercritical fluid chromatography using Chiral Pak IC-3 as the chromatographic column,and determined by atmospheric pressure chemical ionization followed by tandem quadrupole mass spectrometry.The result showed that the detection limit of (+)-methamidophos and (+)-acephate in tea was 5 μg/kg,the limit of quantification was 10 μg/kg.The average recoveries were 70.53%-110.61% and RSD were 2.92%-13.30%.This method can effectively separate and quantify methamidophos and acephate enantiomer.
Key wordsQuEChERS;Supercritical fluids chromatography-tandem mass spectrometry;Tea;Methamidophos;Acephate;Enantiomer
手性是指物质本身立体空间结构左右对称呈镜像,如同人的左右手,相似却不能重叠,是自然界本质属性之一[1]。受生产技术、成本及实际使用等条件的约束,目前世界上有近25%的商用农药具有对映异构体[1],而在我国这一比例估计超过40%[2]。在农药中手性对映异构体常表现出不同的生物活性,一种对映体具有高靶标活性的同时,另一种可能是低效或无效的。另一方面多数手性农药其中一个对映体比其外消旋体显示出更强的急性毒性,且与其余对映体的急性毒性存在显著差异[3]。如腈菌唑的(+)-对映体的抗菌活性是(-)-对映体的1.79~1.96倍,同时其毒性也更大[4];多效唑的杀菌活性(2R,3R)-(+)-对映体高于(2S,3S)-(-)-对映体,但对植物生长调节活性则恰好相反;不同锐劲特的对映体在网纹水蚤体内所表现出的急性毒性[5]和生殖毒性[6]也不同。目前,为保护环境和人体健康,很多国家和地区立法限定外消旋农药的使用,要求必须使用具有正向活性的单一异构体,减少低活性异构体的使用[7-8]。
甲胺磷和乙酰甲胺磷是高效的有机磷杀虫剂,其分子结构中均包含1个磷原子手性中心,分别有2个对映异构体。根据Bertolazzi等[9]的研究,(+)-甲胺磷具有潜在的神经毒性,而(-)-甲胺磷则无法引起神经损伤。乙酰甲胺磷、甲胺磷的(+)-对映体对家蝇的毒性较(-)-对映体更大[10]。(+)-甲胺磷对大型蚤的毒性是(-)-甲胺磷的7倍,而在体外牛细胞试验中,(-)-甲胺磷对乙酰胆碱酯酶的抑制作用比(+)-甲胺磷强8.0~12.4倍[11]。由于甲胺磷毒性强烈,2008年我国就停止对其生产使用,而乙酰甲胺磷虽可使用,但在植物体内会转化成甲胺磷[12]。在我国现行有效的食品中农药残留限量标准中,甲胺磷、乙酰甲胺磷在茶叶中的最大残留限量均为50 μg/kg,但标准仅针对外消旋体含量制定,因此,研究毒性不同的對映体的识别和定量分析技术,有利于更科学地评估其使用过程中的安全性。
近年来,手性农药的拆分及定量分析技术逐渐发展,伴随手性固定相的不断开发,包括液相色谱[13-15]、气相色谱[16-17]、毛细管电泳[18]等分离技术被运用在手性化合物拆分工作中,有报道使用手性液相色谱[19]和气质联用法[20]对甲胺磷或乙酰甲胺磷进行了对映体拆分。超临界流体色谱作为一种新型的分离方法,在手性化合物拆分上有独特的优势[21-22],目前,已在包括有机磷类[23]、拟除虫菊酯类[24]、砜亚胺类[25-26]、三唑类[27-28]、苯氧羧酸类[29]等手性农药的对映体拆分中得到了应用,但鲜见采用超临界流体色谱对甲胺磷或乙酰甲胺磷进行对映体拆分的相关报道。该研究建立了基于QuEChERS前处理技术、超临界流体色谱(SFC)分离、串联质谱法检测的茶叶中甲胺磷、乙酰甲胺磷对映体拆分及定量检测的技术方法,以期为对映体活性研究奠定工作基础。
1材料与方法
1.1试剂药品甲胺磷外消旋体(CAS10265-92-6,纯度99%),购自德国Dr.Erenstofer公司;乙酰甲胺磷外消旋体(CAS30560-19-1,纯度98%),购自美国USP公司;甲醇、异丙醇、乙腈、乙醇和甲酸均为色谱纯,购自美国thermo Fisher公司;二氧化碳(纯度99.99%)、高纯氮(纯度≥99.999%)、高纯氩(纯度≥99.999%),购自北京亚南气体公司;QuEChERS试剂包,购于美国安捷伦科技;试验用水来自Milli-Q纯化水系统,电阻率18.2 MΩ·cm。
1.2仪器设备岛津超临界色谱-串联质谱仪(Nexera UC),配有LC-30ADSF二氧化碳输送泵、LC-20ADXR四元溶剂输送泵、SFC-30A背压调节单元、SIL-30AC自动进样器、CTO-20AC柱温箱、CBM-20A系统控制器,购自日本岛津公司。LCMS-8050三重四极杆质谱仪,配有大气压化学电离源,使用高纯氮气作雾化气和干燥气,高纯氩气为碰撞气,购自日本岛津公司。操作软件为岛津Labsolution v.5.82 SP1。Thermo X1R高速冷冻离心机,购自美国赛默飞世尔科技。水平振荡器,购自德国IKA公司。旋转蒸发仪,购自日本东京理化株式会社。
1.3色谱条件色谱柱为Daicel Chiral-pak IC-3 SFC柱(3.0 mm×150 mm,3 μm);流动相A为超临界二氧化碳,B为异丙醇;梯度洗脱:0~4.0 min 10%~42%B,4.0~6.0 min 42% B,6.0~6.5 min 42%~10% B,6.5~10.0 min 10% B;流速2 mL/min,柱温20 ℃;背压10 MPa,进样体积1 μL,自动进样器温度10 ℃,洗针溶液为乙腈。
1.4质谱条件甲胺磷对映体的质谱参数为母离子142.0(m/z), Q1预四级杆电压-12 V,子离子94.2/125.2(m/z),Q3预四级杆电压-15 /-20 V,碰撞能量-15/-18 eV;乙酰甲胺磷对映体的质谱参数为母离子183.7(m/z), Q1预四级杆电压-14 V,子离子143.2/125.2(m/z),Q3预四级杆电压-25/-20 V,碰撞能量-11/-18 eV;添加剂为1% (V∶V)甲酸甲醇溶液,以流速0.2 mL/min在进入质谱前与色谱馏出液混合。采用大气压化学电离源,正离子模式,多反应监测模式采集。离子驻留时间50 ms,雾化气流速3 L/min,干燥气流速5 L/min,碰撞气压力270 kPa,离子传输管温度150 ℃,加热模块温度200 ℃,接口温度300 ℃。
1.5标准溶液的配制准确称取甲胺磷、乙酰甲胺磷外消旋体各10.0 mg于10 mL容量瓶中,用乙腈定容至刻度,得到浓度为10 mg/mL的外消旋体溶液。由于外消旋体为摩尔比1∶1的对映体混合物,因此在溶液中(+)-甲胺磷、(-)-甲胺磷、(+)-乙酰甲胺磷和(-)-乙酰甲胺磷的浓度均为5 mg/mL。使用前根据需要用异丙醇稀释。
1.6样品前处理茶叶样品均购自本地市场,并保存于干燥通风的室温环境中。样品经粉碎后,称取2.0 g,置于50 mL旋盖离心管中,加入20 mL水充分浸润2 h以上,加入20 mL乙腈、4 g无水硫酸镁、1 g氯化钠、1 g 柠檬酸钠和0.5 g 倍半水合柠檬酸氢二钠,剧烈振摇1 min,在水平振荡器上以300 r/min 的转速振摇15 min以混合均匀。8 000 r/min离心10 min,取12 mL上层清液,置于含1 200 mg MgSO4、400 mg C18、400 mg N-丙基乙二胺(PSA)、45 mg石墨化炭黑(GCB)的净化管中,涡旋振荡5 min,取10 mL上清液,于40 ℃下旋蒸至近干,加入1.0 mL异丙醇溶解残渣,过0.22 μm滤膜,上机测定。
2结果与分析
2.1对映体拆分条件的优化保留因子(k1、k2)、分离因子(α)及分离度(Rs)均用来评价不同色谱条件下对映体拆分的效果。若需要在色谱系统上达到良好的分离度Rs,则需要评估所选用色谱系统中目标化合物的k1、k2值及α值,其中k1、k2值更多与色谱系统的固定相、流动相及温度等条件相关。因此在对映体拆分的优化过程中,对固定相、流动相(改性剂)的选择以及有可能影响化合物色谱保留的SFC背压和柱温条件进行研究。
选择了Chiral-pak IC-3[3.0 mm×150 mm,3 μm,固定相纤维素-三(3,5-二氯苯基氨基甲酸酯),Daicel]、Chiral-pak AD-H[4.6 mm×250 mm,5 μm,固定相直链淀粉-三(3,5-二甲基苯基氨基甲酸酯),Daicel]、Chiral-pak ID-3(3.0 mm×150 mm,3 μm,固定相直链淀粉-3-氯苯基氨基甲酸酯,Daicel)3款手性色谱柱及一款常规反相色谱柱CAPCELL PAK MGⅢ C18(2.0 mm×150 mm,3 μm,固定相C18,Shiseido)作为对比。流动相总流速设置为2 mL/min,分别使用甲醇、乙腈、异丙醇、乙醇作为改性剂,改性剂梯度比例根据不同色谱柱进行调整以达到合适的保留时间(1.5~6.5 min)。對比了甲胺磷对映体和乙酰甲胺磷对映体在相应色谱条件下的分离度Rs。
表1为各柱在不同改性剂条件下的保留因子、分离因子及分离度。在常规反相柱(MGⅢ)上,使用甲醇、乙醇和乙腈作为改性剂时化合物在柱上无保留,且在部分改性剂(异丙醇)条件下甲胺磷和乙酰甲胺磷的对映体不能有效分离(Rs<1)。在使用手性固定相的情况下,在以直链淀粉-3-氯苯基氨基甲酸酯为固定相的ID-3柱以及以直链淀粉-三(3,5-二甲基苯基氨基甲酸酯)为固定相的AD-H柱上,甲胺磷对映体均不能有效分离。在IC-3柱上使用甲醇、乙醇作为改性剂时,乙酰甲胺磷对映体也不能得到分离,对比使用乙腈和异丙醇的情况(图1),在乙腈作为改性剂时,色谱基线较高,杂质峰明显,虽分离度比使用异丙醇时略有提高,但依然不能达到后续工作的要求;在异丙醇作为改性剂时,两对对映体达到有效分离,Rs均不低于1.5,同时基线平稳,无明显干扰峰。因此,选择IC-3手性柱作为分离介质,异丙醇作为SFC改性剂。
在确定了色谱固定相和流动相之后,对同样能够影响色谱保留时间的SFC背压及柱温也进行了优化,这二者在SFC色谱中对超临界二氧化碳(scCO2)的状态起关键作用,进而影响目标物质的保留和选择性。该研究对比了不同背压压力(10、12、15 MPa)及不同柱温(15、20、30、40、50 ℃)的组合对保留和分离的影响。从表2可以看出,在不同背压条件下,不论柱温如何,目标对映体的保留时间变化不明显,同时分离度也没有改变。另一方面,对比不同柱温发现,随着柱温的下降,乙酰甲胺磷对映体的分离度不断增加,在20 ℃时,分离度达到4.0,柱温继续下降至15 ℃,分离度不再增加。这可能是由于二氧化碳的超临界点为31.1 ℃、7.29 MPa,在SFC系统中,若柱温低于31 ℃,柱内CO2是以液态存在,对目标化合物的溶解度下降,此时,对对映体的选择性主要依靠异丙醇与固定相的相互作用,因此分离度增加,在20 ℃以下,柱内CO2已经达到稳定的液态状态,此时,CO2仅作为流动相载体存在,不具有任何色谱洗脱能力,因此温度继续下降,对映体的保留時间和分离度也不再改变;当柱温高于31 ℃时,柱内CO2以超临界流体的状态存在,对甲胺磷和乙酰甲胺磷的溶解度增加,流动相对目标化合物的洗脱能力增强,出峰加快,导致保留时间提前,分离度降低。
2.2对映体洗脱顺序的确定确定了色谱条件后,使用柱后接收的方式,利用标准品进样,制备得到了浓度大于1 μg/mL的单体化合物样品,并使用在线旋光检测器对其旋光度进行分析,判断每个洗脱峰的手性构型。在SFC系统中,在同一离子通道中,对映异构体的洗脱顺序为(+)-甲胺磷早于(-)-甲胺磷、(+)-乙酰甲胺磷早于(-)-乙酰甲胺磷。
2.3质谱条件的优化在电离源的选择上,对比了使用电喷雾电离源和大气压化学电离源时目标化合物的响应情况,结果如图2所示,可见两对对映体在APCI源电离下响应均明显高于ESI源。为了提高目标对映体在质谱内的响应,同时又不影响SFC的色谱分离效果,采用了柱后补偿的方式,加入一定量的添加剂,以促进目标化合物电离,增强响应。由于甲胺磷和乙酰甲胺磷均包含有含N基团,在源内易形成正离子,故对比了不同酸性浓度的添加剂对目标物质谱响应的影响。在0.2 mL/min的流速下,在柱后分别添加0.1%、0.5%、1.0%、2.0%的甲酸甲醇溶液,从图3~4可以看出,随着甲酸甲醇溶液浓度升高,对映体的质谱信号呈上升趋势,在1.0%时达到最大值,之后,酸度继续提高,对质谱信号的增强效果不明显。
使用单针进样的方式对接口温度、离子传输管温度和加热模块温度进行3因素3水平正交试验优化,其中接口温度为300、350、400 ℃,离子传输管温度为150、200、250 ℃,加热模块温度为150、200、250 ℃。优化试验重复3次,计算3次试验平均值来进行结果分析,确保离子源参数优化的准确性。从表3可以看出,接口温度300 ℃、离子传输管温度150 ℃、加热模块200 ℃的组合是最优化的离子源参数。
2.4样品前处理方法的选择及优化QuEChERS法已经被证实为农产品中农药残留前处理的首选方法,该研究根据前期报道,也采用了QuEChERS法作为前处理技术。针对茶叶基质,其主要含有丰富的黄酮、少量淀粉、氨基酸、挥发性物质、有机酸、微量的其他化学成分(如咖啡因、色素)以及有可能被人为或污染而含有的农药残留及污染物等,为了去除这些干扰杂质,对前处理过程中提取剂及净化手段进行了优化。
该试验选择了4种不同配比的脱水剂,分别为Original(4 g MgSO4+1 g NaCl)、CEN(4 g MgSO4+1 g NaCl+1 g Na3Cit+0.5 g 2Na2HCit·1.5H2O)、Vet(4 g Na2SO4+1 g NaCl)、AOAC(6 g MgSO4+1.5 g NaAC),对比了使用乙腈为提取溶剂的情况下不同脱水剂对提取效率的影响,结果如图5所示。从图5可以看出,使用CEN前处理的样品回收率高于其他3者。这可能是因为有柠檬酸缓冲盐的存在,改变了饱和盐溶液的pH,促使甲胺磷及乙酰甲胺磷对映体电离平衡向分子方向移动,从而使更多的目标物溶解进入乙腈提取液中。
在QuEChERS法中,净化剂的选择对于去除杂质、提高目标化合物色谱峰的信噪比至关重要。C18、PSA、GCB是最为常见的净化吸附剂[30]。该试验选择5种不同配比的净化吸附剂,分别为ALN(750 mg MgSO4+150 mg PSA+150 mgC18+150 mg 中性氧化铝)、GCB(200 mg GCB)、GCB+C18(900 mg MgSO4+150 mg GCB+150 mg C18)、0029(1 200 mg MgSO4+400 mg PSA+400 mg C18+45 mg GCB)、High Pigment(900 mg MgSO4+150 mg PSA+45 mg GCB),对比不同净化吸附剂对目标物回收率的影响,结果如图6所示。从图6可以看出,使用0029(1 200 mg MgSO4+400 mg PSA+400 mg C18+45 mg GCB)作为净化吸附剂,4种对映体的回收率均最高。有研究表明,PSA对茶多酚、叶绿素有净化作用,GCB对咖啡因有净化作用[31],都能够将茶叶中有较大影响的干扰物去除,因此,0029吸附剂达到了更好的净化效果,从而降低了基质效应,得到了更好的回收率。这也与Andrade等[32]的研究结果相一致。
2.5方法验证茶叶中甲胺磷和乙酰甲胺磷对映体经由QuEChERS法提取,在以Chiral-Pak IC-3为色谱柱的超临界流体色谱上拆分,大气压化学电离后串聯四极杆质谱法测定。对该定量方法的基质效应、线性、灵敏度、回收率和精密度等指标进行了考察。
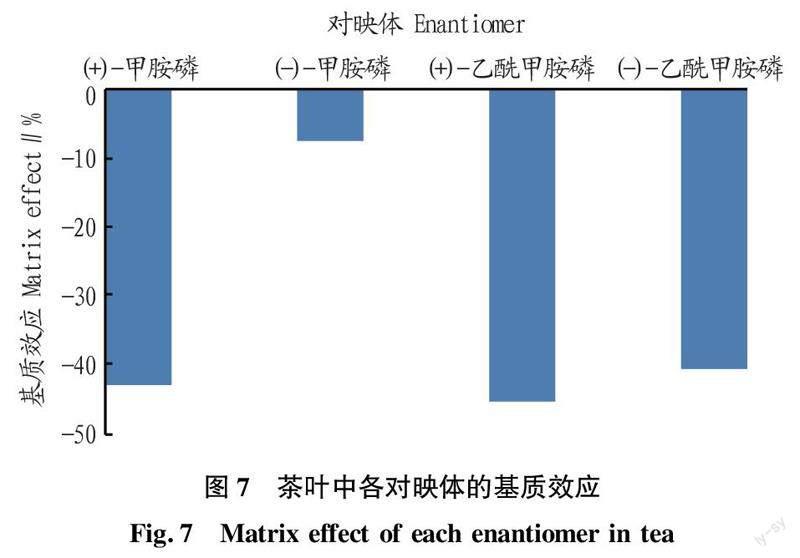
2.5.1基质效应。使用空白茶叶基质,按“1.6”方法进行样品提取、得到空白基质溶液。使用空白基质溶液和异丙醇分别配制浓度为2、5、10、20、50、100 ng/mL的系列基质标准溶液和标准溶液。由茶叶中各对映体的基质效应[33]情况(图7)可知,由于茶叶中基质对电离的干扰较大,4种异构体均表现为基质抑制效应,其中(+)-甲胺磷、(+)-乙酰甲胺磷和(-)-乙酰甲胺磷的基质效应超过40%,基质效应明显,在测定中可选择基质加标标准曲线法定量。
2.5.2标准曲线及灵敏度。配制浓度分别为5、10、20、50、100、200 μg/kg的基质加标标准溶液,以甲胺磷和乙酰甲胺磷对映体的响应峰面积为纵坐标、对应浓度为横坐标绘制工作曲线。以3倍信噪比计算方法检出限(LOD),以10倍信噪比计算方法定量限(LOQ)。(+)-甲胺磷、(-)-甲胺磷、(+)-乙酰甲胺磷、(-)-乙酰甲胺磷的线性方程、决定系数、线性范围、检出限及定量限结果见表4。从表4可以看出,各对映体在5~200 μg/kg线性关系良好,茶叶中4种对映体的检出限和定量限分别为5和10 μg/kg。图8为检出限浓度下加标样品的提取离子流图。
2.5.3回收率与精密度。取空白茶叶样品,根据GB/T 27404—2008的要求,甲胺磷和乙酰甲胺磷均为有最大残留限量(MRL)的化合物,在GB 2763—2016中,茶叶中甲胺磷外消旋体的MRL为50 μg/kg,外消旋体是对映体的等摩尔混合物,因此对于各对映异构体,可看作MRL为25 μg/kg,同样,茶叶中乙酰甲胺磷的外消旋体MRL为 100 μg/kg,对映体MRL为50 μg/kg。 制备浓度分别为10 μg/kg(定量限点)、25 μg/kg(甲胺磷MRL点)、50 μg/kg(乙酰甲胺磷MRL点)3个不同浓度水平的加标样品,在优化后的条件下测定目标化合物,每个浓度水平平行测定6次,结果如表5所示。由表5可知,使用所建立的方法对茶叶中甲胺磷及乙酰甲胺磷对映体进行定量测定,平均回收率为70.53%~110.61%,相对标准偏差(RSD)为2.92%~13.30%。
2.6实际样品测定及方法对比购入市售茶叶15份,涵盖绿茶、花茶、红茶等主要干制茶,按“1.6”方法对样品进行测定。其中某一绿茶样品检出(+)-乙酰甲胺磷21.7 μg/kg、(-)-乙酰甲胺磷19.9 μg/kg,其离子流色谱图见图9,通过阳性样品的离子流色谱图可以看出,乙酰甲胺磷对映体在茶叶上的残留并不会随时间的变化而互相转化,按外消旋体进行判别则此绿茶中乙酰甲胺磷的残留量为41.6 μg/kg。在后续工作中对乙酰甲胺磷对映体的毒性进行进一步研究,可以开展单一对映体农药的使用推广,并进一步针对每种对映体规范限量。
上述阳性样品的检出值与使用GB 中气相色谱法所得到的结果(外消旋体残留量)进行了对比,气相色谱法所得到的结果为39.7 μg/kg。由此可见,所建立的SFC方法不仅试验的环境友好度更高、稳定性更好,同时能在检出值无明显差别的情况下分离对映异构体。
3结论
该研究建立了一种简单、快速、可靠的基于超临界流体色谱-串联质谱法的茶叶中甲胺磷、乙酰甲胺磷对映体拆分及定量检测方法。优化了不同色谱柱、改性剂、柱温和背压等色谱条件,两对对映体达到有效分离,基线平稳,并优化了质谱条件以保证方法灵敏度。在基于QuEChERS法的前处理技术中经优化选择了CEN(4 g MgSO4+1 g NaCl+1 g Na3Cit+0.5 g 2Na2HCit·1.5H2O作为脱水剂,0029(1 200 mg MgSO4+400 mg PSA+400 mg C18+45 mg GCB)为净化吸附剂。经方法确认,方法检出限为5 μg/kg,定量限为10 μg/kg,满足已经制定的MRL检测的需求。同时线性、回收率、精密度等指标均满足相关方法学考察的要求。通过实际样品测试及对比,该方法在检测速度、准确度及环境友好度上具有优势,并且能够有效拆分和定量甲胺磷和乙酰甲胺磷对映异构体,为进一步研究提供方法参考。
参考文献
[1] WILLIAMS A.Opportunities for chiral agrochemicals[J].Pesric Sci,1996,46(1):3-9.
[2] ZHOU Y,LI L,LIN K D,et al.Enantiomer separation of triazole fungicides by high-performance liquid chromatography[J].Chirality,2009,21(4):421-427.
[3] SMITH S W.Chiral toxicology:Its the same thing…only different[J].Toxicol Sci,2009,110(1):4-30.
[4] SUN M J,LIU D H,QIU X X,et al.Acute toxicity,bioactivity,and enantioselective behavior with tissue distribution in rabbits of myclobutanil enantiomers[J].Chirality,2014,26(12):784-789.
[5] OVERMYER J P,ROUSE D R,AVANTS J K,et al.Toxicity of fipronil and its enantiomers to marine and freshwater non-targets[J].J Environ Sci Health Part B,2007,42(5):471-480.
[6] WILSON W A,KONWICK B J,GARRISON A W,et al.Enantioselective chronic toxicity of fipronil to Ceriodaphnia dubia[J].Arch Environ Contam Toxicol,2008,54(1):36-43.
[7] SCHMITT P,GARRISON A W,FREITAG D,et al.Application of cyclodextrin-modified micellar electrokinetic chromatography to the separations of selected neutral pesticides and their enantiomers[J].J Chromatogr A,1997,792(1/2):419-429.
[8] BUERGE I J,POIGER T,MLLER M D.Enantioselective degradation of metalaxyl in soils: Chiral preference changes with soil pH[J].Environ Sci Technol,2003,37(12):2668-2674.
[9] BERTOLAZZI M,CAROLDI S,MORETTO A,et al.Interaction of methamidophos with hen and human acetylcholinesterase and neuropathy target esterase[J].Arch Toxicol,1991,65(7):580-585.
[10] MIYAZAKI A,NAKAMURA T,KAWARADANI M,et al.Resolution and biological activity of both enantiomers of methamidophos and acephate [J].J Agric Food Chem,1988,36(4):835-837.
[11] LIN K D,ZHOU S S,XU C.Enantiomeric resolution and biotoxicity of methamidophos[J].J Agric Food Chem,2006,54(21):8134-8138.
[12] UPADHAY N,KUMAR V,KUMAR V.Extraction,UV-visible,FTIR,NMR spectroscopic study of Acephate and effect of pH [J].Int J Environ Sci,2013,3(6):1849-1856.
[13] KABLER A K,CHEN S M.Determination of the 1′S and 1′R diastereomers of metolachlor and S-metolachlor in water by chiral liquid chromatography-mass spectrometry/mass spectrometry (LC/MS/MS)[J].J Agric Food Chem,2006,54(17):6153-6158.
[14] ZHANG H,WANG X Q,QIAN M R,et al.Residue analysis and degradation studies of fenbuconazole and myclobutanil in strawberry by chiral high-performance liquid chromatography-tandem mass spectrometry[J].J Agric Food Chem,2011,59(22):12012-12017.
[15] ELLINGTON J J,EVANS J J,PRICKETT K B,et al.High-performance liquid chromatographic separation of the enantiomers of organophosphorus pesticides on polysaccharide chiral stationary phases [J].J Chromatogr A,2001,928(2):145-154.
[16] LIU W P,GAN J J.Determination of enantiomers of synthetic pyrethroids in water by solid phase microextraction-enantioselective gas chromatography[J].J Agric Food Chem,2004,52(4):736-741.
[17] WU C W,ZHANG A P,LIU W P.Risks from sediments contaminated with organochlorine pesticides in Hangzhou,China [J].Chemosphere,2013,90(9):2341-2346.
[18] JARMAN J L,JONES W J,HOWELL L A,et al.Application of capillary electrophoresis to study the enantioselective transformation of five chiral pesticides in aerobic soil slurries[J].J Agric Food Chem,2005,53(16):6175-6182.
[19] QIU J,DAI S H,ZHENG C M,et al.Enantiomeric separation of triazole fungicides with 3-μm and 5-μm particle chiral columns by reverse-phase high-performance liquid chromatography[J].Chirality,2011,23(6):479-486.
[20] PAN R,CHEN H P,WANG C,et al.Enantioselective dissipation of acephate and its metabolite,methamidophos,during tea cultivation,manufacturing,and infusion[J].J Agric Food Chem,2015,63(4):1300-1308.
[21]WANG X Y,ZHANG H,XU H,et al.Direct chiral determination of acephate and its metabolite methamidophos in vegetables using QuEChERS by gas chromatography-tandem mass spectrometry[J].Food Anal Methods,2013,6(1):133-140.
[22] NELANDER H,ANDERSSON S,OHLN K.Evaluation of the chiral recognition properties as well as the column performance of four chiral stationary phases based on cellulose (3,5-dimethylphenylcarbamate) by parallel HPLC and SFC[J].J Chromatogr A,2011,1218 (52):9397-9405.
[23] WILLIAMS K L,SANDER L C,WISE S A.Comparison of liquid and supercritical fluid chromatography for the separation of enantiomers on chiral stationary phases [J].J Pharm Biomed Anal,1997,15(11):1789-1799.
[24] WEST C,CIESLIKIEWICZ-BOUET M,LEWINSKI K,et al.Enantiomeric separation of original heterocyclic organophosphorus compounds in supercritical fluid chromatography[J].Chirality,2013,25(4):230-237.
[25] NISHIKAWA Y.Supercritical fluid chromatographic separation of synthetic pyrethroids on packed columns and capillary columns[J].Anal Sci,1992,8(6):817-822.
[26] CHEN Z L,DONG F S,XU J,et al.Stereoselective separation and pharmacokinetic dissipation of the chiral neonicotinoid sulfoxaflor in soil by ultraperformance convergence chromatography/tandem mass spectrometry[J].Anal Bioanal Chem,2014,406(26):6677-6690.
[27] DEL NOZAL M J,TORIBIO L,BERNAL J L,et al.Separation of albendazole sulfoxide enantiomers by chiral supercritical-fluid chromatography[J].J Biochem Biophys Methods,2002,54(1/2/3):339-345.
[28] LIU A,DONG F S,XU J,et al.Stereoselective determination of tebuconazole in water and zebrafish by supercritical fluid chromatography tandem mass spectrometry[J].J Agric Food Chem,2015,63(28):6297-6303.
[29] TORIBIO L,DEL NOZAL M J,BERNAL J L,et al.Chiral separation of some triazole pesticides by supercritical fluid chromatography[J].J Chromatogr A,2004,1046(1/2):249-253.
[30] QI P P,WANG X Y,ZHANG H,et al.Rapid enantioseparation and determination of isocarbophos enantiomers in orange pulp,peel,and kumquat by chiral HPLC-MS/MS[J].Food Anal Methods,2015,8(2):531-538.
[31] CHEN H P,YIN P,WANG Q H,et al.A modified QuEChERS sample preparation method for the analysis of 70 pesticide residues in tea using gas chromatography-tandem mass spectrometry[J].Food Anal Methods,2014,7(8):1577-1587.
[32] ANDRADE G C R M,MONTEIRO S H,FRANCISCO J G,et al.Liquid chromatography-electrospray ionization tandem mass spectrometry and dynamic multiple reaction monitoring method for determining multiple pesticide residues in tomato[J].Food Chem,2015,175:57-65.
[33] ANASTASSIADES M,LEHOTAY S J,STAJNBAHER D,et al.Fast and easy multiresidue method employing acetonitrile extraction/partitioning and“dispersive solid-phase extraction” for the determination of pesticide residues in produce[J].J AOAC Int,2003,86(2):412-431.
