拟天然产物化学进化研究进展
2023-06-26吴广畏高华王莹崔格李姝漩吕锐李德海
吴广畏 ,高华,王莹,崔格,李姝漩,吕锐,李德海*
(1. 南京林业大学化学工程学院,江苏 南京 210037;2. 中国海洋大学医药学院,山东 青岛266100)
天然产物是发现药物先导化合物的重要资源,包括农业上的新型植物生长调节剂芸苔素内酯和赤霉素、农用抗生素井冈霉素和阿维菌素等;畜牧业上真菌来源抗生素截短侧耳素及其衍生物以及70%的人类抗感染药物如棘白菌素等均来自天然产物。特别是因青蒿素、阿维菌素、辣椒素等“诺贝尔奖”分子的贡献,国内外重燃对天然产物挖掘利用的兴趣,说明天然产物及其衍生物在推动人类社会发展中仍然扮演举足轻重的角色且具有巨大潜力。
天然产物极大推动了药物发现,据统计50%以上的药物分子与天然产物关联密切,或提供药效团,或提供模板分子,或受此启发获得灵感。不容忽视的是天然产物直接成药性逐渐下降。导向性应用天然产物(中间体)已经成为科研工作者以及制药企业重要延伸方向,其中创制结构多样化是充分发挥天然产物潜力的重要组成,也是获得药物实体先导的基础。围绕天然产物(中间体)结构多样化的策略多有报道,包括结构多样性导向的合成(diversity-oriented synthesis,DOS)[1]、复杂性到多样性(complexity to diversity,CtD)[2]、生物功能导向的合成(biology-oriented synthesis,BIOS)[3]、功能导向的合成(function-oriented synthesis,FOS)以及最近中科院昆明植物所普诺·白玛丹增团队提出的药效团导向的半合成(pharmacophore-oriented semisynthesis,POSS)等策略[4],这些方法从不同角度对天然产物结构进行了化学演化,发现包括抗新型冠状病毒感染的分子等在内的多个先导化合物。
近年Herbert Waldmann 提出了“拟天然产物”(pseudo-natural product,PNP)化学进化策略(见图1),其核心是利用不同天然产物中存在的片段(fragments)进行组合获得PNP 的结构[5-7]。这些结构可通过化学合成、非酶催化或酶催化获得,为创制新药提供了尚未探索的分子宝藏。更为重要的是PNP 化学空间与美国食品药品监督管理局(Food and Drug Administration,FDA)批准的小分子药物化学空间重叠性高,可能具有更好的成药基础,因此近年来得到了较为广泛的关注。PNP 的设计原则主要包括[8]:1)尽可能形成新的手性中心。手性是许多药物发挥活性的重要基础,天然片段之间通过复杂的组合形成新的三维骨架,包括新手性形成等。2)尽可能有杂原子参与。氮原子、氧原子等杂原子在天然产物和药物分子中的占比非常重要,杂原子的参与有利于获得成药性。3)具有不同生物功能的天然产物片段之间的组合有助于获得新活性的PNP 骨架。4)生物合成上迥异的片段之间组合有助于发现新颖活性(片段不具有的)。原因在于这些片段通过不同的酶合成,具有不同的结构特征,有利于与不同的蛋白结合,从而能发挥新颖生物功能。本文将从化学合成驱动、非酶催化驱动、酶催化驱动等方法组合天然产物片段,形成PNP 的视角概述其发展进程。
1 化学合成驱动的拟天然产物
目前化学合成驱动是制备PNP 的主要方法。Herbert Waldmann 在该领域建树颇丰。PNP 既可通过从头组合(de novocombination)合成,也可直接通过天然片段组合偶联获得。从头组合涉及单个或多个天然产物片段的化学构建,最终获得预设的PNP 结构。直接组合方法中天然片段不需要构建,直接通过化学反应将片段组合成预设的PNP。除了纯天然片段或天然产物中含有的片段,从合成产物(药物)提取的片段与天然产物片段化学偶联生成PNP 的策略有力地扩充了PNP 化学进化策略适用范围,从而获取成药性更高的PNP 类群[9]。
2019 年,Herbert Waldmann 课题组[9]首次阐述PNP 进化策略,并进行了初步验证。色烷(chromane)和苯并吡喃(chromene)片段存在于许多天然产物中,并赋予了这些天然产物多样的生物活性,如结构中有色烷片段的儿茶酸和bitucarpin A 分别具有抗肿瘤和抗细菌活性。同时,天然片段四氢嘧啶酮(tetrahydropyrimidinone,THPM)是许多抗生素的母核,如抗生素TAN-1057A/B 等。Herbert Waldmann 课题组基于从头化学合成法获得了45 个由色烷(苯并吡喃)片段和四氢嘧啶酮片段偶联的chromopynones 化合物(见图2 和3),其代表了一类新的化学结构,该结构类群从生物合成角度很难获得。这些产物所覆盖的化学空间有别于天然产物以及其他合成策略所获得的产物,接近已批准的上市药物,且主要参数符合里宾斯基五规则(Lipinski’s“Rule of Five”,Lipinski-Ro5)。
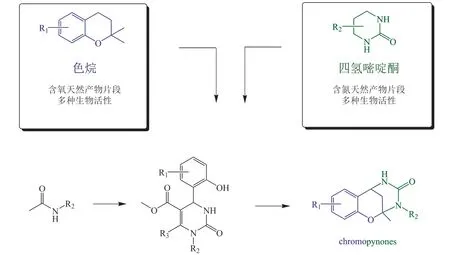
图2 Chromopynones 拟天然产物的设计[9]Figure 2 Design of chromopynone pseudo-natural products[9]

图3 拟天然产物chromopynones 的合成路线和代表性产物[9]Figure 3 Synthetic route and representative products of chromopynone pseudo-natural products[9]
Schneidewind 等[10]利用吲哚和托品烷(tropane)为起始片段从头组合,通过非对映选择性1,3-偶极环加成反应合成了吲哚托品烷(indotropanes)PNP,其中化合物myokinasib 为尚未报道过的新化学类型(见图4)。它可以损害细胞质分裂、诱导多核细胞的形成以及选择性抑制肌球蛋白轻链激酶1(myosin light chain kinase1,MLCK1)的活性,从而降低应力纤维上磷酸化肌球蛋白Ⅱ轻链的丰度,有望成为用于研究MLCK1 介导的细胞进程的独特分子工具[10]。
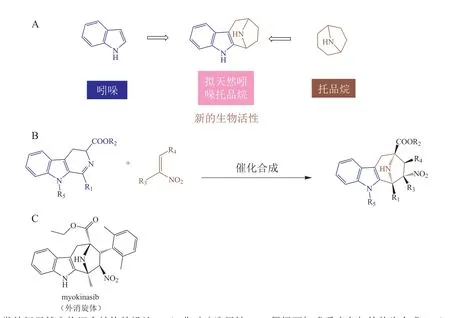
图4 吲哚托品烷的设计原理和合成[10]Figure 4 Design principle and synthesis of indotropanes[10]
吲哚和吗吩烷生物碱是重要活性片段来源。Herbert Waldmann 课题组[11]设计、合成了由吲哚和吗吩烷生物碱组合的indomorphan 类PNPs(见图5A),与常规合成方法获得的产物相比,indomorphans PNPs具有更高度的三维立体特征,进一步地利用Lipinski-Ro5 进行分析发现,61%的indomorphans PNPs 位于类药(drug-like)区域,提示这些PNPs 是药物发现的理想合集。同时,生物活性测试发现这些PNPs 是一类结构新颖的糖摄入抑制剂(活性达纳摩尔级),且仅当吲哚和吗吩烷生物碱偶联之后才会发挥强效的抑制活性,单个片段并未表现出显著的生物活性(见图5B)。其中化合物(±)-glupin 拆分后的单体活性存在显著差异,(+)-glupin 活性更佳。(+)-Glupin可选择性靶向抑制葡萄糖转运蛋白1(glucose transporter 1,GLUT1)和3(GLUT3)活性,从而强效抑制肿瘤细胞的生长[12](见图5B)。
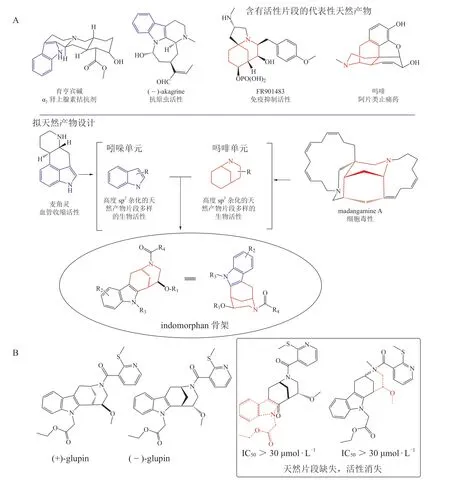
图5 Indomorphans 拟天然产物中天然产物片段的来源以及该类拟天然产物的设计和代表性结构[11]Figure 5 Source of natural product fragments of indomorphans pseudo-natural products & design and representative structures of such pseudo-natural products[11]
很多天然活性生物碱中含有四氢喹啉(tetrahydroquinoline)和四氢吡咯(pyrrolidine)等含氮片段,如可卡因(cocaine),communesin B和virantmycin。Liu 等[12]基于这两大类天然片段,设计了8 种连接方式(见图6),共合成了155 个结构丰富多样的吡咯喹啉(pyrroquinoline,PQ)PNPs。除了连接方式多样外,立体化学重排、不同的饱和度等化学因素丰富了结构多样性。在合成这类结构中,作者还发现了1 个氧化环加成形成喹啉盐(quinolinium salts)的新方法。研究显示,四氢喹啉和四氢吡咯片段形成的不同结构骨架类型具有不同的生物活性。
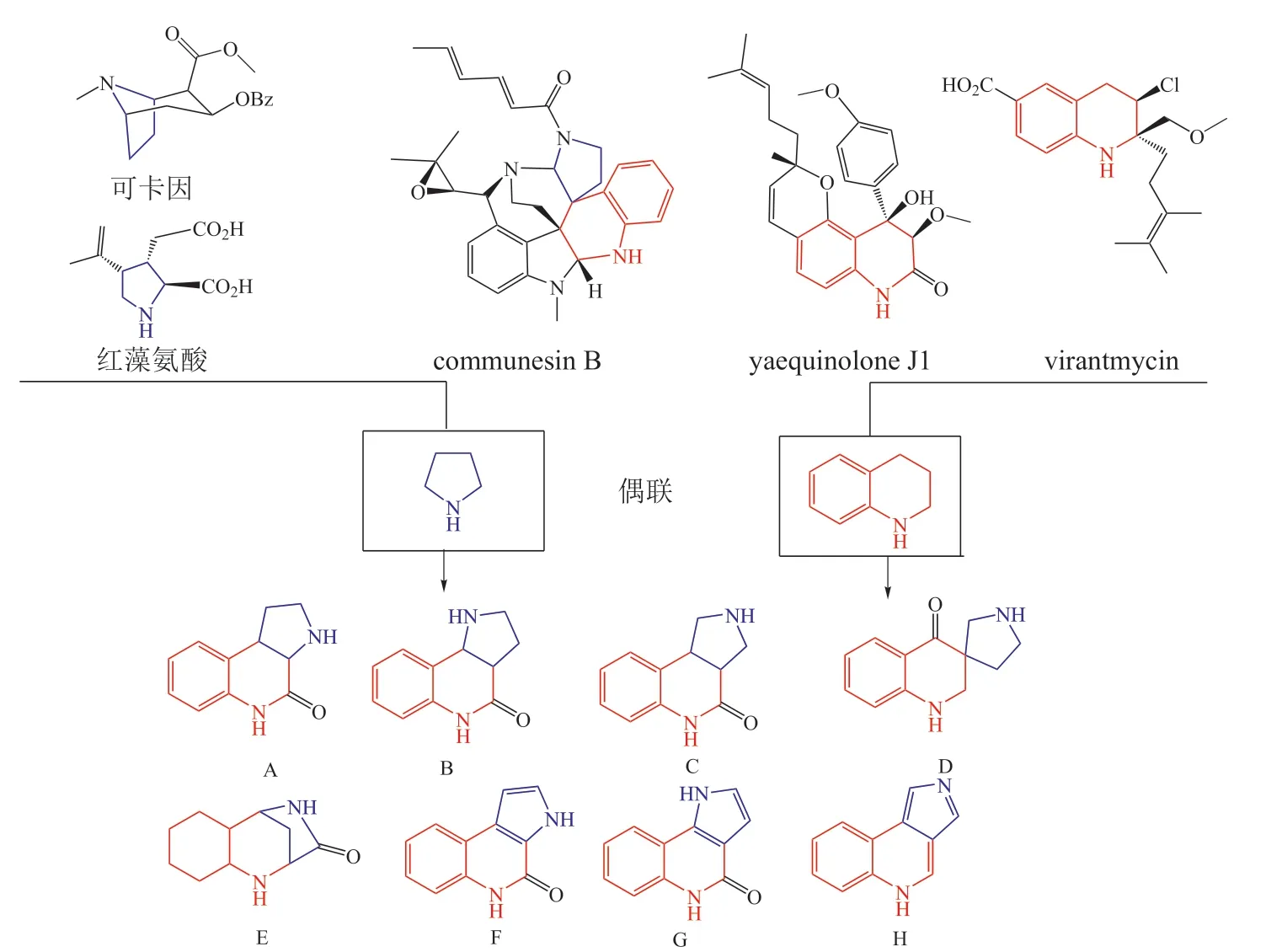
图6 吡咯喹啉拟天然产物的设计[12]Figure 6 Design of pyrroquinoline pseudo-natural products[12]
苯二氮䓬(benzodiazepine)和异吲哚啉酮(isoindolinone)结构单元存在于许多药物分子和天然产物中,不仅是重要的活性母核,而且二者参与多样的天然产物形成,如安曲霉素(anthramycin),sclerotigenin,pestalachloride A,lennoxamine 等(见图7A)。由于这2 个片段富含杂原子,获得的新结构可能具有很好的生物活性,如合成的苯二氮䓬-异吲哚啉酮类PNP MMV000917 可以干扰疟疾寄生虫的离子平衡。Yuan 等[13]利用可回收的介孔二氧化硅纳米粒子(mesoporous silica nanoparticles,MSNs)催化从头合成了21 个四环的苯二氮䓬-异吲哚啉酮类PNPs(见图7B),实验发现MSNs 能够催化多组分反应,收率在55% ~ 91%,其中部分化合物达到了克级制备,且反应过程中无需金属,故能耗低,节能环保,与此同时MSNs 也可以回收再利用。
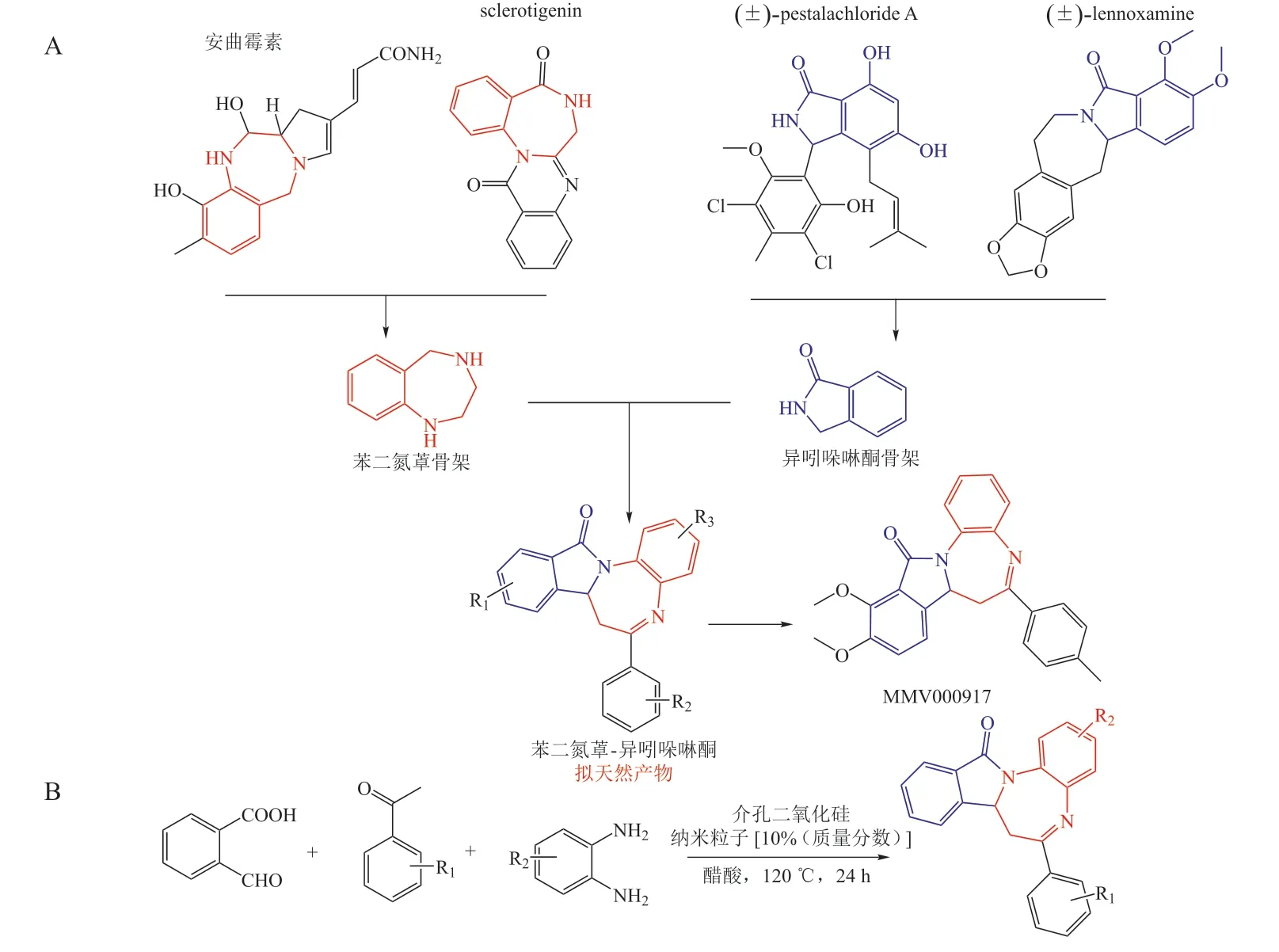
图7 苯二氮䓬-异吲哚啉酮类拟天然产物中天然产物片段的来源以及该类拟天然产物的设计和代表性结构[13]Figure 7 Source of natural product fragments of benzodizepine-fused isoindolinone pseudo-natural products& design and representative structure of such pseudo-natural products[13]
Brosowsky 等[14]以天然产物thailandepsin B 为母核并引入不同的活性弹头,设计合成出一系列thailandepsin B PNPs。其中一些产物表现出强效、选择性的组蛋白去乙酰化酶(histone deacetylase,HDAC)抑制活性。
Burhop 等[15]提出天然产物片段可以通过非生物合成过程中的转化组合形成新颖的PNP。皮克特-施彭格勒(Pictet-Spengler,PS)反应在多环生物碱合成中是重要的转化步骤。类似的oxa-PS 反应被使用在复杂吲哚生物碱合成过程中。作者创新性地使用isomeric-oxa-Pictet-Spengler (iso-oxa-PS)反应,将2-羟乙基吲哚(2-hydroxyethyl indoles)和来源于灰黄霉素(griseofulvin)等天然产物中含有羰基的片段产物组装起来,得到indofulvins PNP 库(见图8)。化学信息学分析表明,indofulvins 结合了已上市药物、类药和天然产物的优良特性。生物活性评价显示,indofulvins 能够调控线粒体功能而抑制自噬现象,提示indofulvins 是一类尚未报道过的、具有新化学类型的自噬抑制剂。
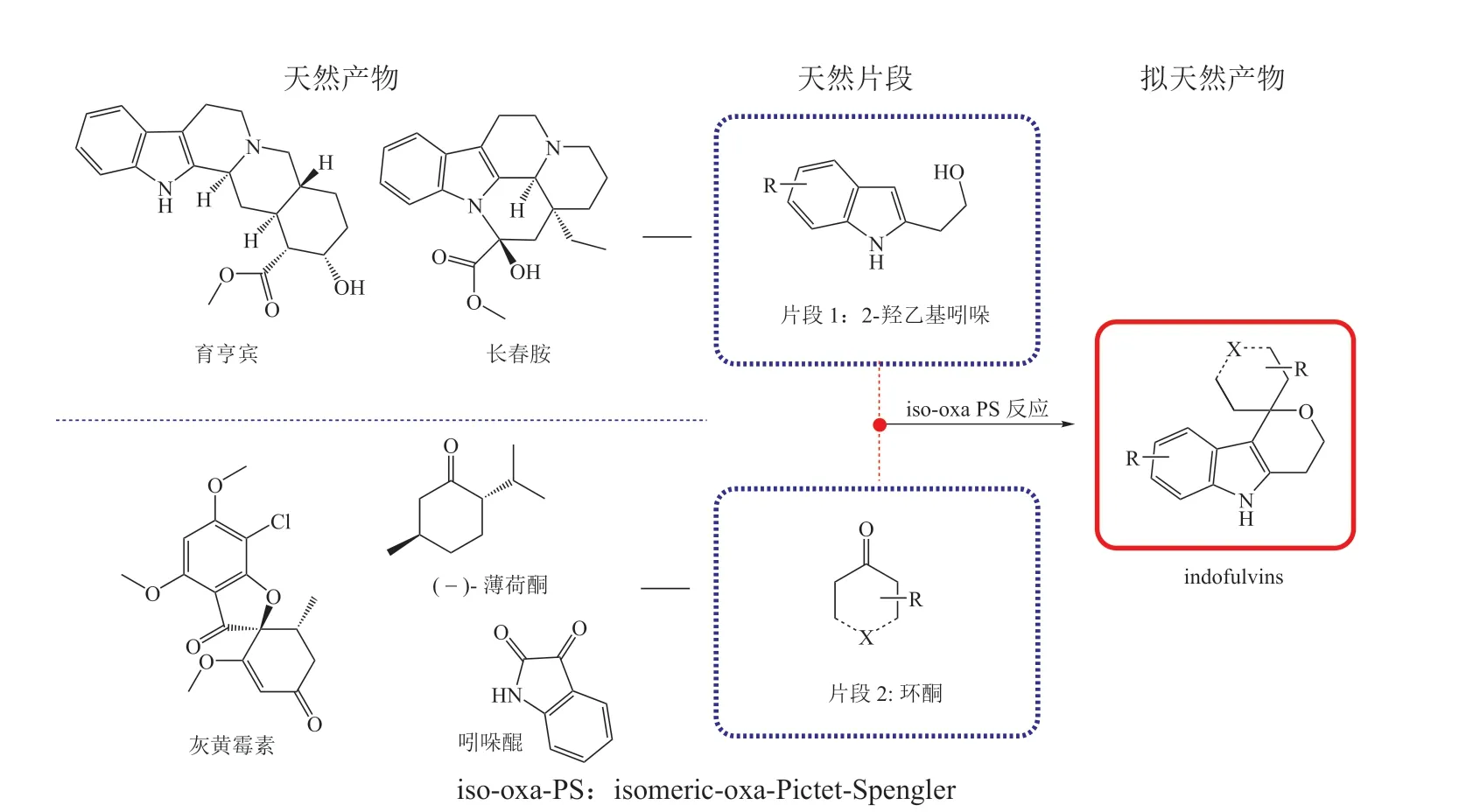
图8 吲哚衍生片段与含有羰基的天然产物片段融合的拟天然产物[15]Figure 8 Pseudo-natural products combining both indole-containing fragments and carbonyl groupscontaining natural product fragments[15]
Davies 等[16]利用吲哚和四氢吡啶片段以单键的形式偶联,产生了一类自然界中不存在的apoxidoles PNPs(见图9)。生物活性评价发现apoxidoles 是一类新的吲哚胺2,3-双加氧酶1(indoleamine 2,3-dioxygenase 1,IDO1) Ⅳ型抑制剂, 该类抑制剂是目前的研究热点。晶体结构分析发现,apoxidoles 能选择性靶向载脂蛋白-IDO1,阻止血红素结合,引起独特的氨基酸定位。
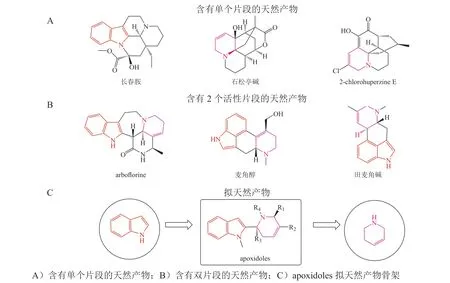
图9 Apoxidoles 拟天然产物的设计[16]Figure 9 Design of pseudo-natural product apoxidoles[16]
天然产物二聚化能够有效地扩展化学空间,且很多研究发现二聚化产物活性比单体活性更强,因此天然产物的聚合是化学进化的重要手段。Niggemeyer 等[17]利用天然产物金鸡纳生物碱,先通过反应将其奎宁环上的双键转化为醛,后通过高效、立体选择性一锅法合成对称和非对称的含有20元环金鸡纳生物碱片段的拟天然生物碱二聚体(见图10)。这些合成的20 元环二聚化产物包括18 个手性中心和1 个额外的天然产物片段。对产物进行进一步的官能化后,衍生获得了163 个大环PNPs。其中化合物tantalosin-I 显著诱导微管相关蛋白1 轻链3(microtubule-associated protein 1 light chain 3,LC3)蛋白的脂化过程,该化合物用于研究LC3 辅助的过程非常有价值。
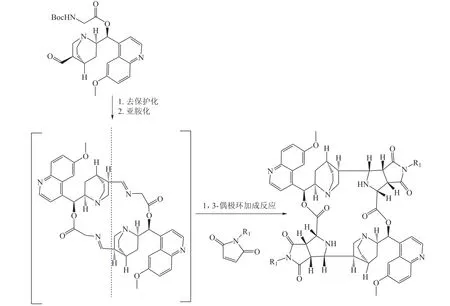
图10 金鸡纳生物碱衍生片段和吡咯烷片段组成的大环拟天然产物的合成[17]Figure 10 Synthesis of macrocyclic pseudo-natural products composed of cinchona alkaloid-derived fragments and pyrrolidine fragments[17]
Herbert Waldmann 课题组[18]对灰黄霉素(griseofulvin)和苯并二氢吡喃酮(chromanone)中的片段进行组合偶联,获得一系列拟天然聚酮化合物(见图11)。生物活性以及化学信息学分析发现化合物grismonone 具有最强的生物活性,其可以显著抑制Hedgehog 信号通路,其作用靶标为Smoothened(SMO)蛋白。Grismonone 代表了新的抑制SMO 化学类型,构效研究发现单独的2 个片段并不能抑制Hedgehog 信号通路,提示片段融合产生了片段所不具备的新生物功能。
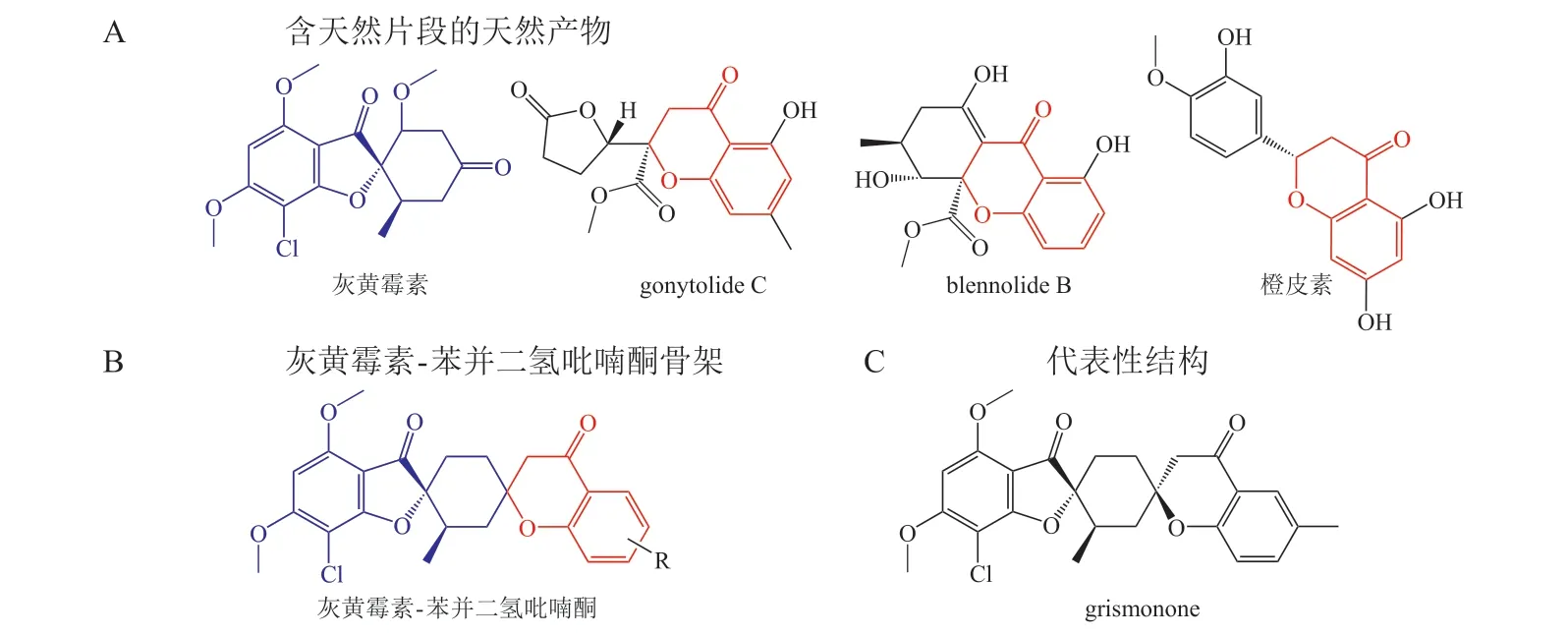
图11 灰黄霉素-苯并二氢吡喃酮拟天然产物中天然产物片段的来源以及该类拟天然产物的骨架和代表性结构[18]Figure 11 Source of natural product fragments of griseofulvin-chromanone pseudo-natural products &skeleton and representative structure of such pseudo-natural products[18]
Zhu 等[19]基于断马钱子苷(secologanin)通过一系列温和的化学反应,包括一步对映选择转化环外的C8 位、C8/C11 位、C8/C9/C10 位,以及化学酶法参与的环内C2/C6 的亲核修饰,合成了一系列PNPs。合成过程中涉及和利用了断马钱子苷中所有可能的反应位点。活性筛选发现,合成所得的化合物vb 具有抗疟疾活性,而化合物IIf 具有高效的神经保护功能(见图12)。

图12 基于断马钱子苷的拟天然生物碱的多样性合成[19]Figure 12 Diversity-oriented synthesis of seclolganin-based pseudo-natural alkaloids[19]
Alonso 等[20]在PNP 合成片段方面进行了拓展。作者依次通过分子内的乌吉(Ugi)反应和皮克特-施彭格勒反应,将合成药物片段和天然甾体偶联成多环的骨架产物(见图13),产物分布在不同的化学空间。
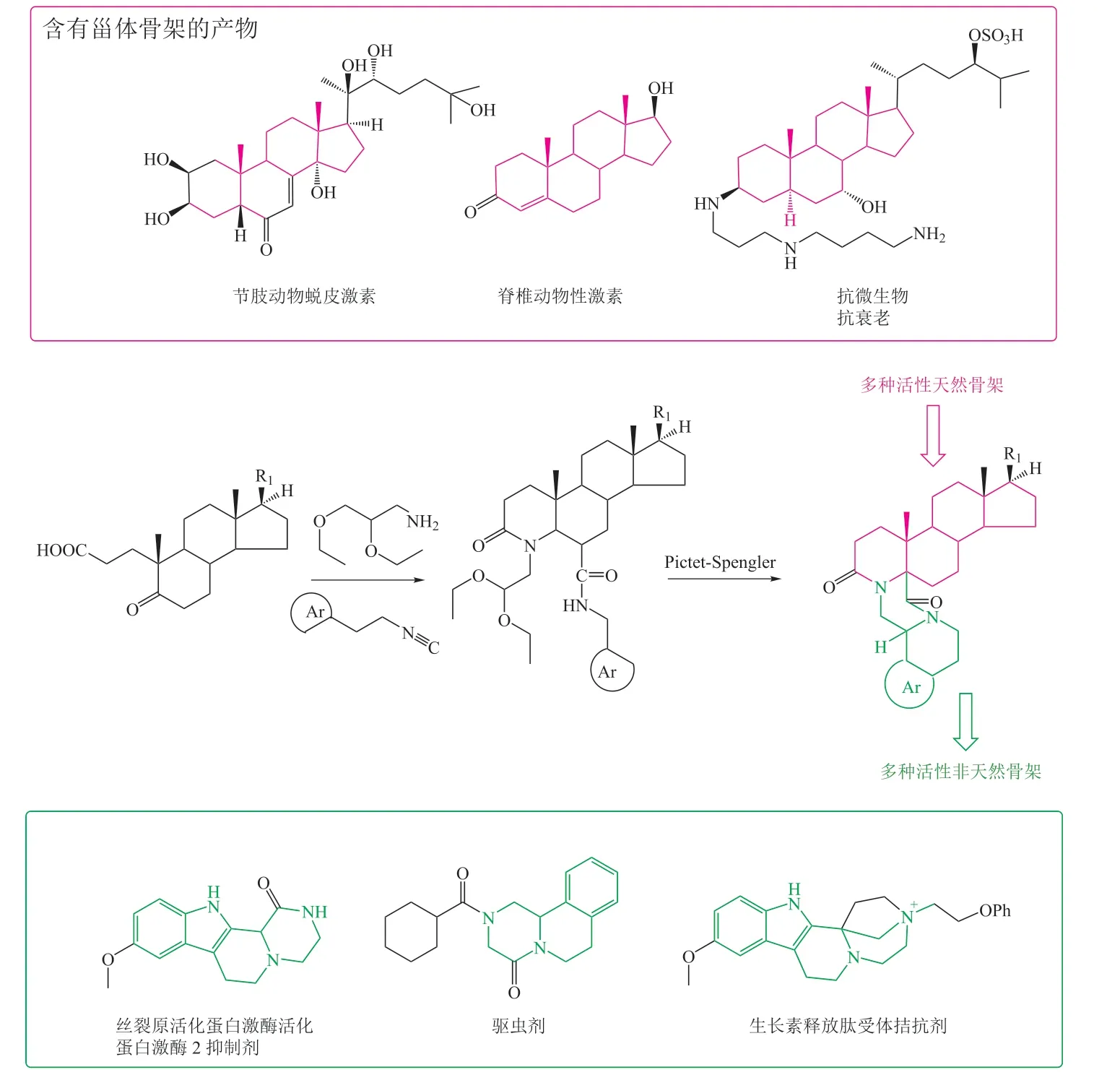
图13 整合天然和非天然片段合成立体化学丰富的多环拟天然产物[20]Figure 13 Synthesis of polycyclic pseudo-natural products with rich stereochemistry by combination of natural and unnatural fragments[20]
2 非酶催化驱动的拟天然产物
随着对天然产物生物合成的不断深入认识,越来越多的证据显示许多复杂天然产物形成过程经常涉及非酶催化反应[21-22]。有趣的是,一些复杂天然产物是由若干天然片段经非酶催化偶联组合形成,这些产物及其非酶催化反应利用生物信息学技术手段难以预测,因此挖掘并利用这些片段和非酶催化反应具有潜在的药源价值,也能揭示天然产物化学进化的过程和生态功能,为PNP 结构设计提供了宝贵资源。
例如,顾谦群、李德海课题组从1 株南极深海来源真菌Penicillium crustosumPRB-2 中分离到新骨架化合物penilactones A 和B,推测其可能是由clavatol 等天然片段经非酶催化的迈克尔加成反应偶联生成[23]。德国马尔堡大学Li Shuming 课题组验证penilactone A 是由3 个片段经过非酶催化的1,4-迈克尔加成反应(1,4-Michael addition)产生,其中clavatol 片段存在ortho-quinone methide(o-QM)等价物[24]。QMs 广泛存在于自然界中,参与了许多天然活性物质的生物合成,是重要的药效基团,也是设计PNP 的理想天然片段。
受penilactones 启发,笔者课题组利用clavatol的o-QM 过渡中间体及其非酶催化性质,在培养P. crustosumPRB-2 过程中添加适当浓度的吲哚等含氮分子,分离鉴定了4 个结构新颖的含clavatol单元的PNPs(见图14),酶失活等实验证实该类结构是通过非酶催化驱动形成的[25]。拟生物碱penindolone 表现出显著的抗禽流感病毒活性,且无明显细胞毒活性。Penindolone 不易产生耐药性。小鼠体内试验表明,喷雾或滴鼻给药都能显著提高小鼠的成活率。作用机制研究发现,penindolone 靶向血凝素(hemagglutinin),能够同时阻止病毒进入细胞以及病毒和细胞之间的膜融合过程,是一类新型的抗病毒先导化合物[25]。
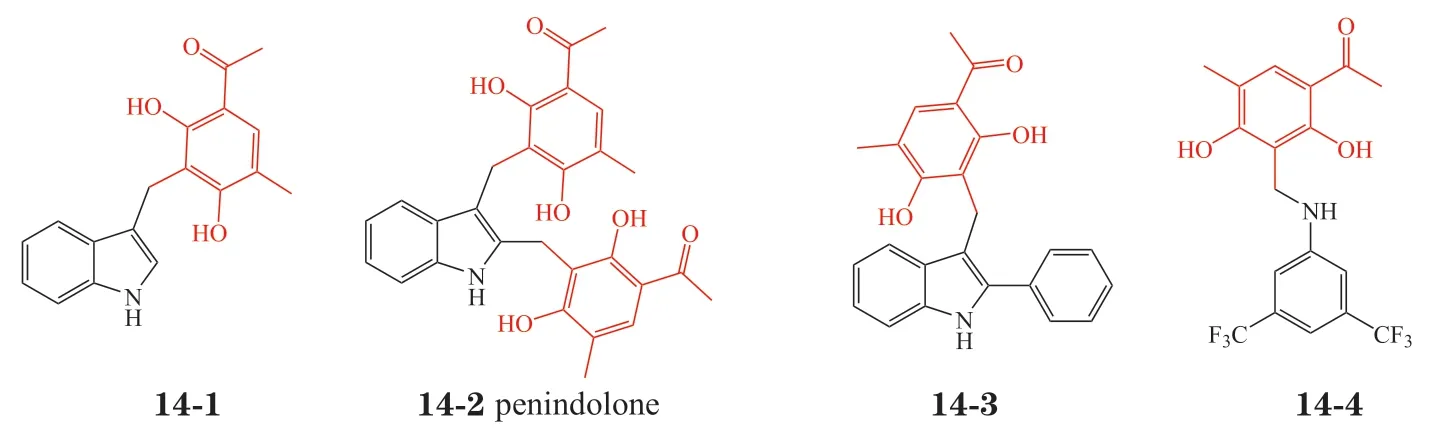
图14 微生物发酵中非酶催化驱动形成的拟天然产物[25]Figure 14 Nonenyzmatic-driven pseudo-natural products formed through microbial fermentation[25]
后续工作中,课题组引入非酶自组装策略,以拓展PNP 结构丰富度。在深入调查P. crustosumPRB-2 产PNP 潜能时,又发现5 种新结构类型的14个PNPs(15-1 ~ 15-14),包括一类高度氧化修饰的氧杂角四环骨架(如化合物15-1 和15-2),该拟天然新骨架分子表现出中等强度的抗肿瘤活性[26]。鉴于该菌产生的活泼中间体及其创造PNP 的潜力,笔者课题组通过将P. crustosumPRB-2 与Xylariasp.共培养,分离鉴定由clavatol 和1,3-dihydroxy-5-(12-hydroxyheptadecyl) benzene 偶联获得的penixylarins A 和B(15-15 和15-16),验证发现这2 个PNPs中的clavatol 片段(见图15 红色部分)是通过非酶催化的迈克尔加成偶联形成[27]。
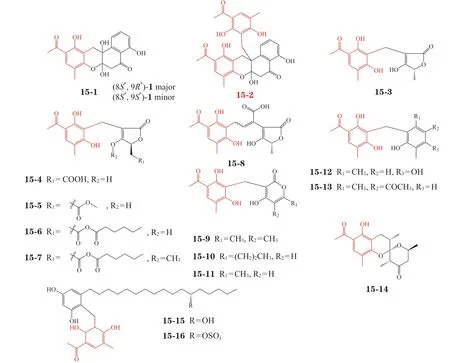
图15 非酶催化驱动发现的内源性含clavatol 拟天然产物[26-27]Figure 15 Endogenous clavatol-containing pseudo-natural products discovered by nonenyzmatic-driven approach[26-27]
2022 年,Purdy 等[28]系统总结了o-QM 参与天然产物生物合成、形成机制和潜在的利用价值。综述中描述了合成o-QM 所需的酶,包括黄素依赖的氧化酶(flavin-dependent oxidases),hetero-Diels-Alderases,S-腺苷-L-甲硫氨酸依赖性周环酶(S-adenosyl-Lmethionine-dependent pericyclases)和α-酮戊二酸依赖的非血红素铁酶(α-ketoglutarate-dependent nonheme iron enzymes)等[28],这些研究为充分利用QM 设计PNP 奠定了酶学基础,对利用合成生物学制备QM产物以及挖掘新的QM 具有重要指导价值。
戴好富课题组和唐啸宇课题组合作研究了血竭(龙血树受伤后产生的分泌物)中查尔酮二聚体形成机制[29],经证实这些二聚化产物是查尔酮的p-quinone methide(p-QM)活泼中间体经非酶促迈克尔加成获得,这些化合物很可能属于PNP 范畴。
除QM 借助非酶促产生PNP,苯胺及其类似物与微生物次级代谢产物偶联也是产生PNP 的理想策略。Zhang 等[30]通过在培养基中添加苯胺类似物,从1 株黄河沼泽来源真菌Penicillim malacosphaerulumHPU-J01 中分离到7 个含苯胺或苯胺类似物的PNPs(见图16),其中6 个为新化合物,化合物talaroenamine F 表现出对蜡样芽胞杆菌(Bacillus cereus)的抗菌活性,最低抑菌浓度(minimum inhibitory concentration,MIC)为0.85 mg · L-1。为了进一步获得结构多样的talaroenamines,该课题组在制备talaroenamine F 的基础上,通过苯胺衍生物的替换,获得利用前体直接添加策略难以制备的19 个新衍生物(见图17),活性评价结果表明talaroenamine F14(17-14)对K562 具有细胞毒活性,半数抑制浓度(half maximal inhibitory concentration,IC50)为2.2 μmol · L-1[31]。实验证实这些新衍生物均为苯胺和环己二酮衍生物通过非酶催化形成,且这2 个片段在生物合成上差别迥异,符合PNP 设计逻辑。
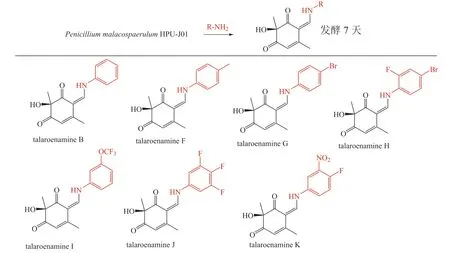
图16 前体直接添加策略合成的含苯胺及其类似物拟天然产物[30]Figure 16 Pseudo-natural products containing aniline and aniline derivatives synthesized by precursor-directed biosynthesis[30]

图17 Talaroenamine F 辅助产生的拟天然产物[31]Figure 17 Pseudo-natural products produced by talaroenamine F-mediated approach[31]
3 酶催化驱动的拟天然产物
利用工程化思路改造核糖体肽生物合成途径是创制拟天然核糖体肽产物的重要手段。目前现有技术编辑核糖体合成和翻译后修饰肽(ribosomally synthesized and post-translationally modified peptides,RiPPs)的生物合成途径存在巨大的挑战。Vinogradov 等[32]基于工程化改造的lactazole A 生物合成途径构建了硫肽(thiopeptides)从头发现的功能平台,通过更换氨基酸类型以及排列组合,可以获得大量的拟天然硫肽产物。结合mRNA 展示技术,可以制备和大规模筛选该平台产生的lactazolelike thiopeptides 拟核糖体肽,结果显示11 个酶促合成的PNPs 中,有9 个对Traf2-和Nck-相互作用激酶(Traf2- and Nck-interacting kinase,TNIK)表现出高亲和力;有10 个抑制了TNIK 活性。进一步研究发现这些拟天然硫肽能够像多肽Tat 一样有效地进入到细胞的胞液中。随着对天然产物生物合成的不断认知,该策略将会成为发现PNP 的重要方向。
4 化学和生物合成联合驱动的拟天然产物
生物合成中的化学活泼中间体、不稳定产物(亦或等价替代物)已经展示了其在拓展结构空间方面的优势。尤其是具有可再生性的微生物源化学活泼物质(中间体),其在很大程度上解决了原料供给的瓶颈。化学活泼中间体具有多向性的化学反应属性,是制备PNP 的理想起始原料。受真菌Chaetomiumsp.产生的chaetophenols 启发,Asai 等[33]推测该类结构可能存在共同聚酮来源的片段,将该聚酮片段的生物合成基因簇pksCH-2构建到米曲霉(Aspergillus oryzae)表达系统中,使转化株大量产生该片段,在Aspergillus oryzae中内源酶的作用下产生o-QMs 活泼等价物。作者基于该活泼的聚酮片段,通过微生物发酵过程中的自身聚合以及与外源片段进行化学合成偶联,制备获得了41 个PNPs,包括聚酮低聚物(polyketide oligomers),azaphilone-type 分子和indole-polyketide 杂合化合物等20 个不同的骨架类型(见图18)。其中1 个化合物表现出显著的抗腺病毒活性,最大效应半数浓度(half-maximum effective concentration,EC50)为4.6µmol · L-1。这种联合策略驱动制备PNP 方式为高效利用微生物中高活性的化学活泼片段(产物)提供了范例。

图18 基于真菌非还原型聚酮合酶途径的拟天然聚酮化合物的结构多样性[33]Figure 18 Structural diversity of pseudo-natural polyketides based on fungal non-reducing polyketide synthase pathways[33]
李德海课题组利用稳定的o-QM 等价物与6 个不同的底物一步合成了15 个PNPs(见图19),其中4 个PNPs 同样可以由非酶催化体获得(见图14),其余11 个PNPs 则难以利用微生物发酵过程中非酶催化获得[25]。
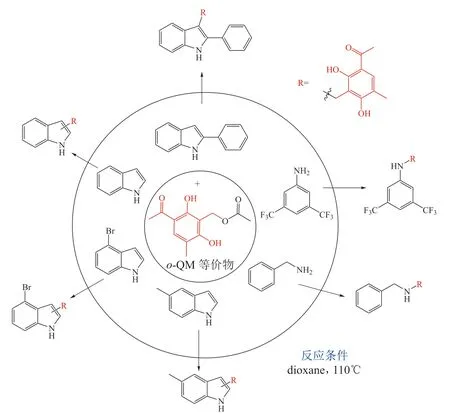
图19 基于clavatol 拟天然产物的结构及其化学合成[25]Figure 19 Structures and chemical synthesis of clavatol-based pseudo-natural products[25]
5 展望
PNP 化学进化策略为高效发现创新性药物小分子提供了重要思路,方兴未艾,充分展示了天然产物药效团在成药性中的优势,并提供了重要的技术手段。天然产物片段的合理选择及设计组合是构建PNP 的关键。事实上,多数天然产物片段供给量难以满足PNP 合集构建,这些片段往往无法直接从天然产物中大量制备获得,这在很大程度上阻碍了PNP 的创制。从头合成是解决其原料来源瓶颈问题的思路之一。重要天然活性物质(植物、微生物来源)生物合成途径的深入研究及其形成机制的阐明,为今后利用合成生物学手段高效制备天然活性物质提供重要信息。微生物来源的化学活泼中间体(产物)参与的PNP 在药物先导物发现方面备受关注,虽然这些化学活泼的产物所参与构建的结构、化学反应难以预测,但是其为设计合成PNP 结构多样性提供了难得的、可再生的天然片段材料。深入研究这些片段也对挖掘微生物的代谢潜能及其生态功能具有重要意义。
