Zonarol 类天然混源萜的合成及其生物活性研究进展
2023-06-20孙盛鑫李圣坤
孙盛鑫, 王 侠,2, 李圣坤*,,2
(1.贵州大学 绿色农药与农业生物工程国家重点实验室培育基地/教育部重点实验室,贵阳 550025;2.南京农业大学 植物保护学院,南京 210095)
天然产物作为自然界中动物、植物、昆虫以及微生物的组成成分或其在适应环境过程中所分泌的代谢产物,拥有独特的结构和丰富的生物学性质,在新型医药和农药的开发中发挥重要作用[1]。其中,萜类化合物作为最大的一类植物次代谢产物,在农业上被广泛应用于病虫草害的防治,如芳樟醇,柠檬烯以及香芹酮等[2]。Drimane 醌/氢醌类化合物是由drimane 型萜类骨架通过杂合生源途径与苯基、取代的苯基或氧化的苯基 (醌) 组成[3],这两部分在结构上通过C(sp3)杂化的末端碳与C(sp2)杂化的芳环组合在一起 (图式1)。在药物化学中,富含C(sp3)杂化的化合物比含C(sp2)杂化的化合物具有更好的药代动力学(PK)和物理化学性质[4],因此C(sp2)-C(sp3)偶联反应及其在药物先导发掘中的应用引起广泛关注[5]。化合物zonarol和isozonarol 是Fenical 等于1973 年从太平洋收集的棕色海藻Dictyopteris zonarioides中首次分离得到的,之后他们用CrO3将zonarol 和isozonarol 氧化得到了zonarone 和isozonarone[6]。1984 年,Kakisawa 等成功地从美国加利福尼亚湾收集的褐藻Dictyopteris undulat中分离得到了zonarone 和isozonarone[7],结构式见图式1。在被分离之初zonarol 表现出抗真菌活性,后续被证实具有拒食活性、抗肿瘤活性、抗炎活性和杀藻活性。本课题组前期对zonarol 及其相关天然产物进行了合成,其抗真菌活性测定结果初步证实zonarol 具有作为新型农药分子模型的潜力[8]。尽管drimane 醌/氢醌类天然产物具有丰富的药理学性质,但是面临着结构复杂和来源有限等问题,使其药理和农用化学潜力未得到充分研究和探索。本文就zonarol及其类似物的合成、结构优化及生物活性方面的研究进展进行阐述,旨在为基于drimane 醌/氢醌骨架的药物先导研发奠定基础并提供借鉴。

图式1 ( ± )-zonarol 及其类似物的结构Scheme 1 The structure of ( ± )-zonarol and its analogues
1 Zonarol 的生物活性、作用机制及药理作用
天然产物zonarol 是drimane 醌/氢醌类中代表性化合物,在其被分离后,其天然类似物isozonarol、zonarone 和isozonarone 也被陆续报道。生物活性和药理学研究表明,zonarol 及其类似物在农业上具有抗菌、杀藻以及拒食等活性。除此之外,在抗肿瘤、抗氧化、免疫调节以及神经保护等方面也有一定的应用。
1.1 抗菌活性
1973 年,Fenical 等从太平洋收集的棕色海藻D.zonarioides中首次分离出zonarol 和isozonarol,并发现其对樟疫霉菌Phytophthora cinnamomi、立枯丝核菌Rhizoctonia solani、核盘菌Sclerotinia sclerotiorum和齐整小核菌Sclerotium rolfsii都表现出中等程度的抑真菌活性[6,9]。1986 年,Mori 等合成了(+)-zonarol 和(-)-zonarol 并测试了其对6 种真菌的抑菌活性,发现(+)-zonarol 及其对映体对新型隐球菌Cryptococcus neoformans和酿酒酵母Saccharomyces cerevisiaeATCC 9763 表现出一定的抑制活性,但对热带念珠菌Candida tropicalisIFO 0006、烟曲霉菌Aspergillus fumigatus、烟曲霉菌Asp.Niger IFO 4417 和须毛癣菌Trichophyton mentagrophytes均没有抑制活性[10]。值得注意的是,两种对映体在所测定的生物活性方面并无差别[10]。2006 年,Joshi等发现,zonarol 对海鱼分枝杆菌Mycobacterium marinum表现出良好的抑制作用[11]。2018 年,本课题组报道了(-)-zonarol 和(-)-isozonarol 及(-)-zonarone和(-)-isozonarone 的混合物对立枯丝核菌和核盘菌表现出中等的抗真菌活性,结果见表1[8]。

表1 (-)-zonarol、(-)-isozonarol、(-)-zonarone 和(-)-isozonarone 的抗真菌活性[8]Table 1 Antifungal activity of (-)-zonarol, (-)-isozonarol,(-)-zonaroneand (-)-isozonarone[8]
1.2 杀藻活性
以α-亚麻酸作为阳性对照,Ishibashi 等评估了zonarol 及其类似物在1 μg/mL 质量浓度下对4 种代表性赤潮微藻种,即赤潮异弯藻Heterosigmaakashiwo、海洋卡盾藻Chattonella marina、古老卡盾藻Chattonella antiqua和长江口水域异甲藻Heterocapsa circularisquama的杀藻活性 (表2)[12]。其中zonarol 和isozonarol 对赤潮异弯藻和长江河口水域异甲藻显示出良好的杀藻活性,isozonarol在1 μg/ml 下对长江河口水域异甲藻的抑制率高达100%;zonarol的类似物(-)-yahazunol、(+)-zonaroic acid 以及(-)-chromazonarol 也表现出良好的杀藻活性。他们探究了zonarol 及其类似物的结构-活性关系,发现当 (+)-zonarol 的C-18 位羟基替换为羧基后,杀藻活性显著降低。(+)-zonarol可以被视为(-)-yahazunol的脱水产物 (结构式见图式2)。从表2 中可以看出,(-)-yahazunol 的杀藻活性优于(+)-zonarol,(+)-zonarol 的杀藻活性与其环化产物(-)-chromazonarol相当[12]。由此得出zonarol 及其类似物的结构与杀藻活性的关系(图式2):1) C-8 位羟基对杀藻活性至关重要;2) 苯环上取代基对杀藻活性有影响,其中羟基的活性优于羧基;3) Drimane 与氢醌之间形成吡喃结构对杀藻活性的影响不大。

表2 Zonarol 及其类似物在1 μg/mL 的杀藻活性[12]Table 2 Algicidal activity of zonarol and its related analogues at 1 μg/mL [12]
1.3 拒食活性
Kurata 等用纤维素板法在每个样品为75 μg 的剂量下测试了化合物的拒食活性。结果表明,化合物zonarol、isozonarol、zonarone 及isozonarone 对幼鲍鱼皱纹盘鲍Haliotis discus表现出较强的拒食活性,选择性指数 (EI):zonarol = 0.85, isozonarol =0.78, zonarone = 0.92, isozonarone = 0.85[13]。
1.4 抗肿瘤活性
Laube 等发现,(+)-zonarol、(+)-zonarone 和(+)-isozonarol 对肿瘤细胞系小鼠成纤维细胞系(L-929) 和人类白血病细胞系 (K-562) 显示出良好的细胞毒性 (表3),此外(+)-zonarol 和 (+)-isozonarol 还对人类宫颈癌细胞系 (HeLa) 表现出良好的抗肿瘤活性[14]。Barrero 等在合成zonarol 相关天然产物的基础上,对ent-isozonarol、ent-isozonarone和ent-chromazonarol 的抗肿瘤活性进行了测定,发现ent-isozonarol 和ent-isozonarone 对肿瘤细胞系P-388、A-549、HT-29 和MEL-28均表现出良好的抑制作用,其中ent-isozonarol 对4 种肿瘤细胞系抑制作用的IC50值均小于1 μmol/L[15](表4)。

表3 (+)-zonarol、(+)-zonarone、(+)-isozonarol 和(+)-isozonarone 对肿瘤细胞系L-929、K-562 和HeLa 的细胞毒性活性[14]Table 3 Cytoxic activity against the tumour cell lines L-929, K-562 and HeLa of (+)-zonarol, (+)-zonarone,(+)-isozonarol and (+)-isozonarone[14]
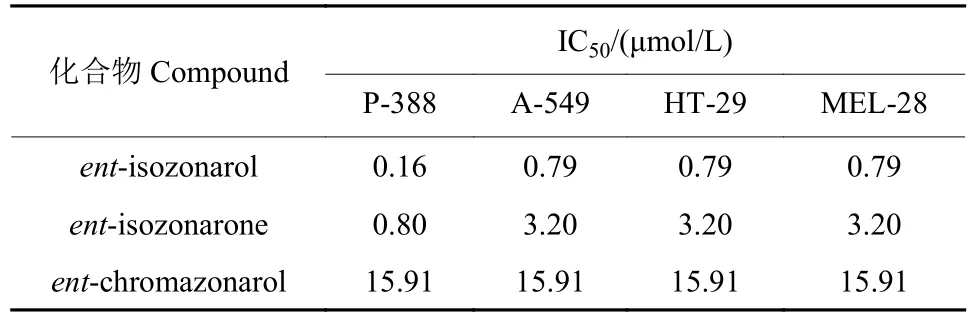
表4 ent-isozonarol、ent-isozonarone 和 ent-chromazonarol对肿瘤细胞系P-388、A-549、HT-29 和MEL-28细胞毒性活性的IC50 值[15]Table 4 The IC50 values of ent-isozonarol, entisozonarone and ent-chromazonarol against tumour cell lines P-388, A-549, HT-29 and MEL-28[15]

图式2 Zonarol 及其类似物的杀藻结构活性关系Scheme 2 The structure and algicidal activity relationship of zonarol and its analogues
1.5 其他生物活性
为了寻找天然源的抗氧化剂,Kumagai 等发现,波纹网翼藻D.undulata的甲醇提取物具有较强的1,1-二苯基-2-苦艾肼(DPPH)自由基清除活性。研究表明,(+)-isozonarol 对DPPH 自由基清除活性强度最高,其 EC50值 (71 μmol/L) 与阳性对照α-生育酚 (EC50= 74 μmol/L) 相当[16],而(+)-isozonarol的氧化产物(+)-isozonarone (EC50= 145 μmol/L) 的抗氧化活性相对于(+)-isozonarol 有所减弱[17]。
3α-羟类固醇脱氢酶(3α-HSD)是炎症级联反应的关键酶之一,Laube 等对(+)-zonarol 及其类似物进行了抗炎活性测定,发现(+)-zonarol 和(+)-isozonarone 对 3α-羟类固醇脱氢酶(3α-HSD)有良好的抑制作用[18]。Yamada 等发现,(+)-zonarol在改善葡聚糖硫酸钠 (DSS) 诱导的结肠损伤方面发挥了关键的作用[19]。Han 等研究并发现,(+)-zonarol可以明显降低炎症细胞浸润、肝脂质沉积和氧化应激[20]。
Shimizu 等发现,(+)-zonarone 能够激活Nrf2/ARE 通路,诱导抗氧化酶,并保护神经元细胞[21]。高浓度的谷氨酸可通过抑制胱氨酸内流消耗细胞内谷胱甘肽(GSH) 来诱导细胞死亡,(+)-zonarol 在1 μmol/L 浓度下几乎完全阻止了谷氨酸诱导的HT22 细胞死亡,其ED50(保护作用)值为0.22 μmol/L[22]。
此外,zonarol 可以作为NO 生成抑制剂[23],还可作为脂肪酶抑制剂,有应用于医药、食品和化妆品行业的潜力[24]。
2 Zonarol 及其类似物的合成
围绕drimane 骨架与醌/氢醌片段的C(sp2)-C(sp3)链接键的构建,zonarol 所代表的drimane 醌/氢醌类化合物的合成策略主要包括Michael 加成反应、芳基金属试剂与羰基的亲核加成反应、Stille偶联和多烯环化反应、硼杂香紫苏内酯合成、Barton脱羧偶联反应及以脱羧硼化和Suzuki 偶联反应等。
2.1 以Michael 加成反应为关键步骤合成
Welch 等最早开展了对(±)-zonarol 的全合成[25]。他们以化合物9 为起始原料,经过12 步以总收率13.8%得到(±)-zonarol[26]。化合物9 经过Wolff-Kishner 还原得到三甲基十氢化萘醇10,然后经琼斯氧化和甲基锂的亲核加成得到三甲基十氢化萘醇12,之后高温脱水得到烯烃中间体13,中间体13 通过间氯过氧苯甲酸(m-CPBA) 处理后,以98% 的产率进行环氧化得到环氧化合物14。用2-异丙基氨基锂处理化合物14,实现环氧开环,接着用Collins 试剂氧化烯丙醇混合物,可得到化合物15。化合物15 与2,5-二甲氧基苯基溴化镁格氏试剂发生Michael 加成反应,实现drimane 与芳香环键连,然后用乙酸酐(Ac2O)处理,得到烯醇乙酸酯,再经氢氧化钾水解便可得到化合物16。化合物16 经过Wittig 反应得到zonarol 的二甲醚17,最后利用正丁基硫醇锂脱甲基得到天然产物(±)-zonarol (图式3)。其中化合物16 进行甲基锂处理后,再经二甲基亚砜区域选择性脱水可得到zonarol 的甲醚混合物(±)-zonarol 17 Δ8,12和 (±)-isozonarol 18 Δ7,8(1 : 4.8),最后脱甲基便可得到天然产物(±)-isozonarol(图式3)。该路线虽然步骤冗长繁琐,但可为后续合成(±)-zonarol 及其相关天然产物提供参考。

图式3 (±)-zonarol 和 (±)-isozonarol 的首次合成[25]Scheme 3 First synthesis of (±)-zonarol and (±)-isozonarol[25]
尽管Welch 等完成了zonarol 的首次合成,但是它的绝对构型仍是未知的。随后,Mori 等完成了zonarol 的两种对映体的全合成,并证实了天然产物(+)-zonarol 的绝对构型[10]。
Mori 等[10]对zonarol 的合成主要分为两个阶段,首先是关键中间体20 的合成,如图式4 所示。以香叶基丙酮yeranylacetone 19 为起始原料,经过3 步反应,以27.05%的产率合成了关键中间体β-羟基酯(±)-20[27]。用(R)-1-(1-萘基)乙基异氰酸酯对(±)-20 进行衍生化,生成了化合物21 和22 的混合物,两个非对映体可以通过柱层析轻松分离。最后用三氯硅烷(HSiCl3) 处理氨基甲酸酯21 和22,分别得到纯化合物(-)-20 和(+)-20。
Mori 工作的第二阶段就是将中间体20 的对映体转化为zonarol 的对映体。首先用氢化铝锂还原(-)-20 得到二醇中间体23,再用对甲基苯磺酰氯(p-TsC1) 处理得到酰化产物24,用琼斯试剂将其氧化得到25,化合物25 的绝对构型经ORDCD 图谱分析得到证实。用DBU 处理25 得到消除产物(-)-15,随后与2,5-二甲氧基苯基溴化镁格氏试剂发生Michael 加成实现了C(sp2)-C(sp3)的键连,然后通过Wittig 反应、去甲基化得到天然产物(+)-zonarol,(+)-zonarol 再经三氧化铬氧化即可得到(+)-zonarone。从香叶基丙酮出发经过12 步以2.6%的产率得到(+)-zonarol。根据相同操作处理(+)-15,可得到(-)-zonarol (总收率2.0%) 和(-)-zonarone。

图式4 Zonarol 对映体的合成[10]Scheme 4 Synthesis of zonarol enantiomers[10]
Akita 等基于二醇(-)-23 实现了(+)-zonarol的立体选择性合成[28]。首先通过(±)-23 和2-羟基-1-萘甲醛反应得到了缩醛,然后以近乎100%的收率实现乙酰化并得到了相应的乙酸酯(±)-26;再通过酰化酶I 对化合物(±)-26 进行立体选择性的水解,得到水解产物(+)-27 和未反应的(+)-26,对其催化氢化即可得到中间体二醇(+)-23 和(-)-23。在催化量的硫酸存在下用苯甲醛处理中间体(-)-23、高产率地获得缩醛28,使用氢化铝锂-三氯化铝还原体系制备了伯醇29。通过四溴化碳和三苯基膦处理,将29 通过Appel 反应转化为溴代物30;然后在Pd(OH)2/C 催化氢解脱除苄基、得到溴醇31。溴醇31 氧化得到溴酮中间体32;用1,8-二氮杂二环十一碳-7-烯(DBU)处理后使其发生消除反应得到所需的α,β-不饱和酮中间体(-)-15。在碘化亚铜作用下,以(-)-15 与2,5-二甲氧基苯基格氏试剂进行Michael 加成实现了C(sp2)-C(sp3)的键连,然后用乙酸酐处理,得到烯醇醋酸盐,其水解为酮(-)-16。在正丁基锂的存在下,(-)-16 与鏻盐发生Wittig 反应得到了烯烃(-)-17,再根据Mori 的方法即可得到(+)-zonarol (图式5)。该合成路线立体选择性强,但由于反应步骤较多而影响了目标化合物的总收率。
2.2 以芳基金属试剂与羰基的亲核加成反应为关键步骤合成
亲核加成反应是另一种常用的获取drimane 醌/氢醌类化合物的方法。该策略包括将醛作为倍半萜前体与芳基锂试剂偶联。1999 年,Barrero 等以(-)-香紫苏醇((-)-sclareol)为起始原料高效合成了ent-isozonarol、ent-zonarol 及ent-isozonarone;并在此基础上通过简单的酸诱导的亲核环化高产率地获得ent-chromazonarol[15](图式6)。首先用OsO4将香紫苏醇氧化为醛33,然后与合成的芳基锂金属试剂进行亲核加成反应以总收率91%高效合成了关键中间体34。中间体34 再经过氯化亚砜的处理,以97%的总收率得到了两类烯烃异构体35a 和35b 的混合产物。中间体35a 和35b 经过多步转化可高效制备一系列zonarol 相关类似物,包括ent-isozonarol、ent-isozonarone、entisozonarone 以及ent-chromazonarol。

图式5 从(±)-23 合成(+)-zonarol[28]Scheme 5 Synthesis of (+)-zonarol from (±)-23[28]
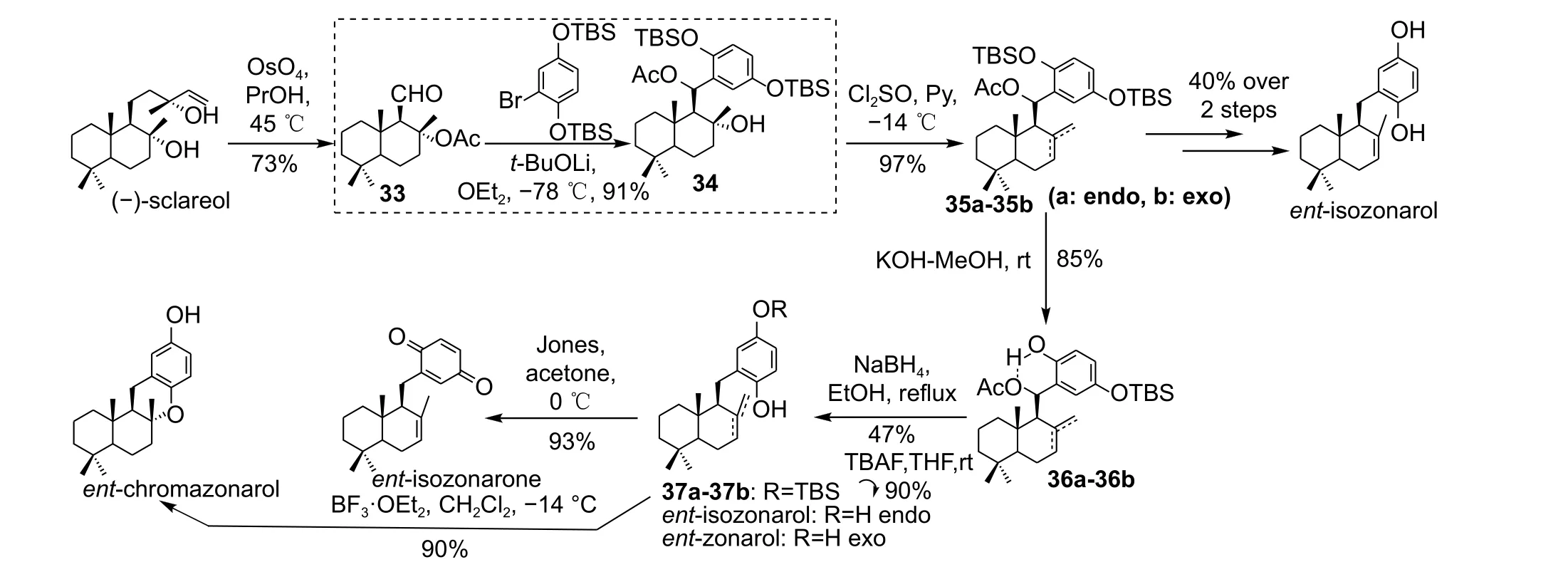
图式6 从(-)-香紫苏醇合成ent-isozonarol、ent-zonarol、ent-isozonarone 和ent-chromazonarol[15]Scheme 6 Synthesis of ent-isozonarol, ent-zonarol, ent-isozonarone and ent-chromazonarol from (-)-sclareol[15]
2000 年,Schröder 等以β-紫罗兰酮 (β-ionone)为起始原料,通过将其转化为关键中间体(+)-albicanal 和(+)-drim-7-en-11-al,从而实现(+)-zonarol、(+)-zonarone、(+)-isozonarol 和(+)-isozonarone 的制备[29]。从β-紫罗兰酮出发合成两种手性中间体醛(+)-44 和(-)-47 的路线如图式7所示。在Wilkinson’s 催化剂存在下,使用Et3SiH将β-紫罗兰酮转化为二氢-β-紫罗兰酮38,然后与碳酸二甲酯进行克莱森缩合得到单环酮酯39,接着使用四氯化锡(SnCl4)将其环化得到化合物(±)-40。(±)-40 经过Wittig 反应、水解反应得到外消旋酸(±)-42。将酸(±)-42 与手性α-苯乙胺混合,通过乙醇的多次重结晶纯化从而实现对映异构体的有效拆分[30]。(+)-42 定量转化为甲酯(+)-41,在Pd/CaCO3和三苯基膦作用下将其异构化生成甲酯(-)-45。用二异丁基氢化铝(DIBAH)将两种酯(+)-41 和(-)-45 分别还原为相应的醇(-)-albicanol(-)-43 和(+)-drim-7-en-11-ol (+)-46。用氯铬酸吡啶(PCC) 氧化得到所需的醛(+)-albicanal (+)-44 和(-)-drim-7-en-11-al (-)-47。
在(+)-zonarone 和(+)-isozonarone 的合成中最重要的一步是倍半萜部分与芳烃单元的偶联。在仲丁基锂条件下,(+)-albicanal (+)-44 和(-)-drim-7-en-11-al (-)-47 分别与芳基锂化合物发生亲核反应实现了C(sp2)-C(sp3)的键连。该反应虽然可高产率得到苄醇48 和50,但是它们是非对映异构体的混合物。利用Li/NH3/NH4Cl 体系进行11 位的去羟基化可定量地得到49 和 51,再经脱保护即可得到(+)-zonarol 和(+)-isozonarol,最后用硝酸铈铵(CAN)进行氧化,得到所需的 (+)-zonarone 和(+)-isozonarone (图式8)。

图式7 合成(+)-albicanal 和(-)-drim-7-en-11-al[29]Scheme 7 Synthesis of (+)-albicanal and (-)-drim-7-en-11-al[29]

图式8 合成zonarol、isozonarol、zonarone 和isozonarone Scheme 8 Synthesis of zonarol, isozonarol, zonarone and isozonarone
随后在2002 年,Laube 等改进了zonarol 和zonarone 的合成,提升了倍半萜与芳烃链接的产率,并在此基础上完成了yahazunol 的制备[31]。该路线先用CAN/吡啶-2,6-二羧酸N-氧化物对(+)-44 进行氧化、脱甲基处理,得到(+)-zonarone,再用连二亚硫酸钠还原成(+)-zonarol。利用苄基保护zonarol 的酚羟基,再用m-CPBA 处理苄基醚54,得到环氧化物55 的非对映体混合物。用氢化铝锂进行环氧开环得到56 (12%) 和57 (56%)。化合物57 脱苯甲酰基保护,即可得到(-)-yahazunol(图式9)。

图式9 合成(+)-zonarol 和 (-)-yahazunol[31]Scheme 9 Synthesis of (+)-zonarol and (-)-yahazunol[31]
同年,Villamizar 等报道了以二萜(+)-迈诺醇((+)-manool)为起始原料合成 (-)-zonarol 和 (-)-zonarone的方法[32]。采用的核心策略与Seifert 课题组之前的报道相似,不同的是制备关键合成子(-)-albicanal(-)-44 的效率大大提高,所需步骤从之前的10 步缩短到4 步 (图式10)。

图式10 从(+)-迈诺醇合成(-)-zonarol 和 (-)-zonarone[32]Scheme 10 Synthesis of (-)-zonarol and (-)-zonarone from (+)-manool[32]
随后,Villamizar 等又以二烯58 作为中间体合成了isozonarol Δ8,9和isozonarone Δ8,9(图式11)[33]。首先二烯58 在m-CPBA 作用下进行环氧化,再用硅胶负载的硼氢化锌裂解环氧化物59,最后用TPPA 或者PCC 氧化即可得到(-)-bicyclofarnesal 63。在正丁基锂存在下,芳基锂化合物加成到醛63 上,以此为关键步骤成功得到苄醇64。苄醇64 经过还原、氧化即可得到isozonarone Δ8,966,将其用连二亚硫酸钠还原便可得到isozonarol Δ8,967。
2006 年,Villamizar 等在完成(-)-zonarol 制备的基础上合成了 (+)-chromazonarol[34](图式12)。首先(+)-迈诺醇经过高锰酸钾氧化成酮68[35], 在氧气存在下,用叔丁醇钾氧化裂解酮68 得到酸69,用氢化铝锂还原酸,随后用四丙基高钌酸铵(TPAP)氧化醇,得到醛70,醛70 经过Baeyer-Villiger氧化得到酯71,生成的甲酸酯用氢氧化钾水解、再用四丙基高钌酸铵氧化后便可得到关键中间体(-)-albicanal (-)-44。在仲丁基锂条件下,醛(-)-44与芳基锂化合物发生亲核加成反应,实现C(sp2)-C(sp3) 的键连,合成了异构苄醇60。用Li/NH3/NH4Cl 还原苄醇 60,再经脱保护即可得到(-)-zonarol,接着利用三氟化硼乙醚将其环化便可得到 (+)-chromazonarol。

图式11 合成isozonarol Δ8,9 和isozonarone Δ8,9[33]Scheme 11 Synthesis of isozonarol Δ8,9 and isozonarone Δ8,9[33]
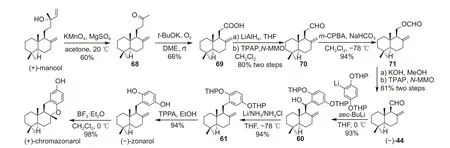
图式12 合成(-)-zonarol 和 (+)-chromazonarol[34]Scheme 12 Synthesis of (-)-zonarol and (+)-chromazonarol[34]
2.3 以Stille 偶联和多烯环化反应为关键步骤合成
2006 年,Gansäuer 等利用Stille 偶联和多烯环化作为关键步骤完成了(-)-zonarol 和(-)-zonarone的全合成 (图式13)[36]。从香叶基香叶醇 (geranylgeraniol)出发合成关键中间体72[37-38],然后与芳基锡烷偶联得到环氧烯烃73。在Cp2TiCl2催化下化合物73 进行多烯环化,然后再脱羟基得到(+)-17,之后用CAN 氧化得到(-)-zonarone,最后用Na2S2O4还原即可得到(-)-zonarol。
2014 年,Garai 等从乙酸金合欢酯 (farnesyl acetate) 出发,仅通过6 步反应便合成了(-)-zonarone[39](图式14)。首先,乙酸金合欢酯在氯磺酸处理下进行多烯环化,得到二环drimane 衍生物75,中间体75 通过在碱性条件下水解,以总产率90%得到drimanol 类化合物76 和77 (物质的量比1 :1),最后中间体76 再经过与芳基金属试剂反应和氧化反应高产率得到(-)-zonarone。
2.4 通过硼杂香紫苏内酯合成
2012 年,Dixon 等描述了通过硼-香紫苏内酯模块化地合成drimane 醌/氢醌类天然产物的方法[40]。如图式15 所示,化合物83 是从香紫苏内酯出发通过5 步以59% 的产率得到的,其中包括,(+)-香紫苏内酯的还原、碘代、消除、水解和硼氢化。在K2S2O8和AgNO3的条件下,通过用过量的1,4-苯醌处理83,实现了C(sp2)-C(sp3)的键连,得到了(+)-chromazonarol。以(+)-chromazonarol为中间体可制备(-)-zonarone、(-)-isozonarone、(-)-zonarol、(-)-isozonarol 和isozonarol Δ8,9,这些化合物的合成比以前的方法更简洁,且产物具有多样性。
2.5 以Barton 脱羧偶联反应为关键步骤合成

图式14 通过乙酸金合欢酯合成(-)-zonarone[39]Scheme 14 Synthesis of (-)-zonarone from farnesyl acetate[39]
2018 年,本课题组从(-)-香紫苏醇出发,以Barton 脱羧偶联作为关键步骤,以最短步骤合成天然产物(+)-yahazunol[8]。合成途径如图式16 所示:在乙酸酐存在下,用高锰酸钾氧化降解(-)-香紫苏醇,以中等收率得到的8-乙酰基drimanic 酸84,酸84 与2-巯基吡啶氮氧化物在Steglich 酯化条件下缩合得到氧化还原活性酯85。在光照条件(250 W) 下,实现活性酯与苯醌的Barton 脱羧偶联、从而完成了C(sp2)-C(sp3)的键连,合成了化合物86。在室温下,用雷尼镍处理化合物86 便可方便快速得到 (+)-yahazunol 的乙酸酯87,之后用氢化铝锂进行还原便可得到(+)-yahazunol。接着用MnO2氧化(+)-yahazunol 后得到(+)-yahazunone。用SOCl2/Et3N处理(+)-yahazunone,得到 (-)-zonarone和(-)-isozonarone 混合物。此外反应的温度影响(-)-zonarone 和(-)-isozonarone 的生成比例。用Na2S2O4还原即可得到(-)-zonarol 和(-)-isozonarol。
该路线与通常将drimanyl 醛与有机锂化合物偶联或“硼香紫苏内酯”偶联的方法相比更加简短,但同样只能得到(-)-isozonarol 和(-)-isozonarol及它们氧化产物的混合物。
2.6 以脱羧硼化和Suzuki 偶联反应为关键步骤合成

图式15 从(+)-香紫苏内酯合成(-)-zonarol 和(-)-zonarone[40]Scheme 15 Synthesis of (-)-zonarol and (-)-zonarone from (+)-sclareolide[40]

图式17 通过Suzuki 偶联合成(-)-zonarol 相关天然产物Scheme 17 Formal synthesis of (-)-zonarol related natural products by Suzuki coupling
本课题组最近开发了脱羧硼化与Suzuki 偶联相结合的一种策略,利用该方法可模块化地合成drimane 醌/氢醌类天然产物及其类似物[41-42]。如图式17 所示,以(-)-香紫苏醇为原料经过三氯化钌和高碘酸钠氧化得到酸84,与N-羟基邻苯二甲酰亚胺缩合生成氧化还原活性酯88。在乙酰丙酮酸铜催化下,化合物88 进行脱羧硼化得到关键合成子89,随后在Pd2(dba)3和RuPhos 组成的催化体系下,高效地实现与溴代芳烃的Suzuki 偶联反应,实现C(sp2)-C(sp3)的键连,以87%的产率得到苄基保护的(+)-yahazunol 90。化合物90 经过脱保护得到(+)-yahazunol[14],再环化便可得到(+)-chromazonarol[8]。根据之前的文献报道的方法即可得到(-)-zonarol 相关天然产物。相比于Baran 小组的方法,该策略合成的关键中间体步骤少、中间体稳定且有良好的化学选择性,可以方便快速地制备天然产物类似物库。
3 总结与展望
本文对zonarol 及其相关天然产物的分离、生物活性及合成研究进展进行了总结,以期为药物新先导的发掘提供很好的参考。关于zonarol 的合成策略及其生物活性概述如图式18 所示,其中合成方法包括α,β-不饱和酮与格氏试剂的Michael 加成、醛与芳香金属试剂的亲核反应、多烯环化、Barton 脱羧偶联及Suzuki 偶联等。
就目前所报道的全合成效率而论,其中羰基化合物与芳香金属试剂的加成反应路线大都较为冗长,并多采用贵金属试剂或易燃催化剂;仿生环化步骤虽短,但只能合成特定的几个天然产物,多样性不足;因此采用C(sp2)-C(sp3)偶联的策略是高效模块化合成zonarol 氢醌类天然产物的有力工具。科学家们正在探索新的高效、经济的合成方法,来替代传统的高成本偶联策略。以zonarol 为代表的drimane 混源萜类化合物表现出丰富的生物学性质,包括抗菌活性、杀藻活性、拒食活性、抗肿瘤活性、抗氧化活性、抗炎活性和神经保护作用等,为药理学研究提供良好基础。此外,手性药物的生物活性与其立体构型密切相关,在drimane醌/氢醌类天然产物中含有多个手性中心,其相关的对映异构体和非对映异构体也被报道,但并未涉及zonarol 及对映异构体之间的构效关系,其手性对其生物活性及作用机制的影响值得进一步研究。相信随着高效且多样性合成方法的开发,必将大力促进从drimane 醌/氢醌类化合物中挖掘全新的农药和医药先导化合物,促进药物先导研究。

图式18 zonarol 合成的化学策略及其生物活性Scheme 18 Chemical synthesis strategies and biological activities of zonarol
