指纹图谱结合一测多评法评价马鞭草质量的方法学研究
2023-06-16刘松照刘丛彬王国凯谢冬梅
王 诤,刘松照,刘丛彬,王国凯,3,谢冬梅,3,4*
(1.宁国市市场监督管理局,安徽 宁国 242300;2.安徽中医药大学 药学院,安徽 合肥 230012;3.中药研究与开发安徽省重点实验室,安徽 合肥 230012;4.安徽省中医药科学院 中药资源保护与开发研究所,安徽 合肥 230012)
马鞭草(Verbenae Herba)为马鞭草科(Verbenaceae)植物马鞭草的干燥地上部分,始载于《名医别录》,原产于热带美洲[1],其味苦、性凉,具有活血散瘀、利水退黄、解毒、截疟的功效[2-3]。马鞭草中主要指标性成分包括环烯醚萜苷类的马鞭草苷、戟叶马鞭草苷以及苯丙素类的毛蕊花糖苷[4-6]。目前有关马鞭草环烯醚萜苷类及苯丙素苷类成分含量测定的报道较少,难以全面控制并评价马鞭草药材的质量。中药指纹图谱能够较全面地反映中药的质量特性,近年来常被用于中药的质量评价[7]。本研究通过建立马鞭草的指纹图谱,结合主成分分析、聚类分析等方法对试验数据进行处理,旨在为马鞭草的质量评价提供科学理论依据。同时由于多指标成分的含量测定中对照品的需求较高,本研究建立了马鞭草中3种指标性成分的一测多评法[8],并与外标法进行比较,证明所建立的方法简便、可靠,可节省大量对照品[9],提高检测效率,现将研究过程报道如下。
1 仪器及材料
1.1 仪器
Shimadzu LC-10A高效液相色谱仪(LC-10ATVP泵,SPD-10AVP紫外检测器,SIL-10ADVP自动进样器,LC-Solution色谱工作站,日本岛津),Agilent C18色谱柱(250mm×4.6mm,5μm),十万分之一电子天平AB135-S(瑞士梅特勒托利多)。
1.2 材料
甲醇、乙腈为色谱纯,水为超纯水;马鞭草苷、戟叶马鞭草苷、毛蕊花糖苷对照品均由本实验室自制,纯度均达98%以上。不同来源的马鞭草药材16 批,见表1。

表1 马鞭草样品16批的产地信息
2 方法与结果
2.1 对照品溶液制备
分别精密称取戟叶马鞭草苷、马鞭草苷及毛蕊花糖苷对照品7.16、11.62、15.47mg,置于10mL容量瓶中,加80%甲醇溶解并稀释至刻度,摇匀;取1mL对照品溶液置于10mL容量瓶中,加80%甲醇溶解并稀释至刻度,摇匀,制成每1mL含0.071 6mg戟叶马鞭草苷、0.116 2mg马鞭草苷和0.154 7mg毛蕊花糖苷的对照品储备液。
2.2 供试品溶液制备
取药材粉碎过80目筛,取粉末约0.5g,精密称定,加80%甲醇25mL,置具塞锥形瓶中,称定重量,超声(功率100W,频率40kHz)提取30min,冷却至室温。80%甲醇补足重量,摇匀,滤过,取续滤液,即得。
2.3 色谱条件
采用Agilent C18色谱柱(4.6mm×250mm,5μm),检测波长为238nm,柱温30℃;流动相:0.1%磷酸水溶液(A)-乙腈(B),体积流量1mL/min,进样量10μL,洗脱程序见表2。在上述色谱条件下,戟叶马鞭草苷、马鞭草苷和毛蕊花糖苷色谱峰分离效果较好,见图1。
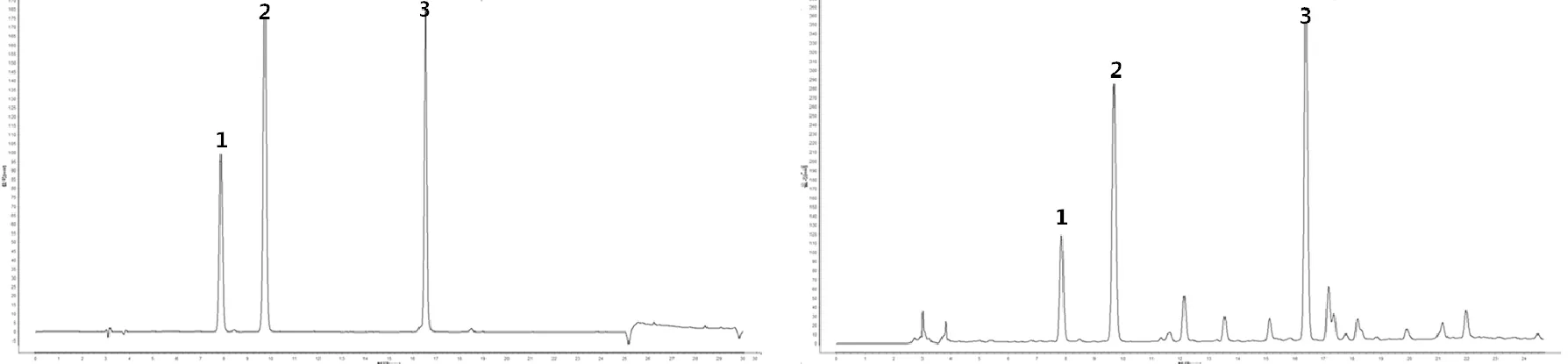
注:1.戟叶马鞭草苷;2.马鞭草苷;3.毛蕊花苷。

表2 梯度洗脱程序
2.4 指纹图谱建立
2.4.1 精密度试验 取S1号样品,按上述方法制备供试品溶液,按“2.3”项下色谱条件测定,连续进样6次,以2号色谱峰(马鞭草苷)的保留时间和峰面积作为参考,计算各共有峰的相对保留时间和相对峰面积,结果其RSD值均<1.5%,6次采集的指纹图谱的相似度>0.98,表明该仪器和所用方法的精密度良好。
2.4.2 稳定性试验 取S1号样品,按上述方法制备供试品溶液,按“2.3”项下色谱条件测定,分别于0、2、4、6、8、10、12h检测,以2号色谱峰(马鞭草苷)为参照峰,测得各共有峰的相对保留时间和相对峰面积,并计算RSD值。结果表明,RSD均<1.5%,且6次采集的指纹图谱的相似度>0.97,表明该仪器和所用方法的稳定性良好。
2.4.3 重复性试验 取S1号样品6份,按上述方法制备6份供试品溶液,并按照“2.3”项下色谱条件测定,以2号色谱峰(马鞭草苷)为参照峰,测得各共有峰的相对保留时间和相对峰面积,并计算RSD值。结果表明,RSD均<1.5%,且6次采集的指纹图谱的相似度>0.97,表明该仪器和所用方法的重复性良好。
2.4.4 特征指纹图谱的建立 应用“中药色谱指纹图谱相似度评价系统(2012版)”对16批马鞭草药材的色谱进行分析,色谱见图2,相似度评价结果见表3。共确定11个共有峰,其中1号峰为戟叶马鞭草苷、2号峰为马鞭草苷、6号峰为毛蕊花苷。经分析可知,11个共有峰保留时间的RSD均<1%,峰面积RSD值在18.33%~81.22%之间。结果显示不同批次的马鞭草化学成分种类相似,但部分成分的含量存在较大差异,见表4、表5。

图2 马鞭草16批的指纹图谱

表3 相似度计算结果

表4 马鞭草共有峰的保留时间 (min)
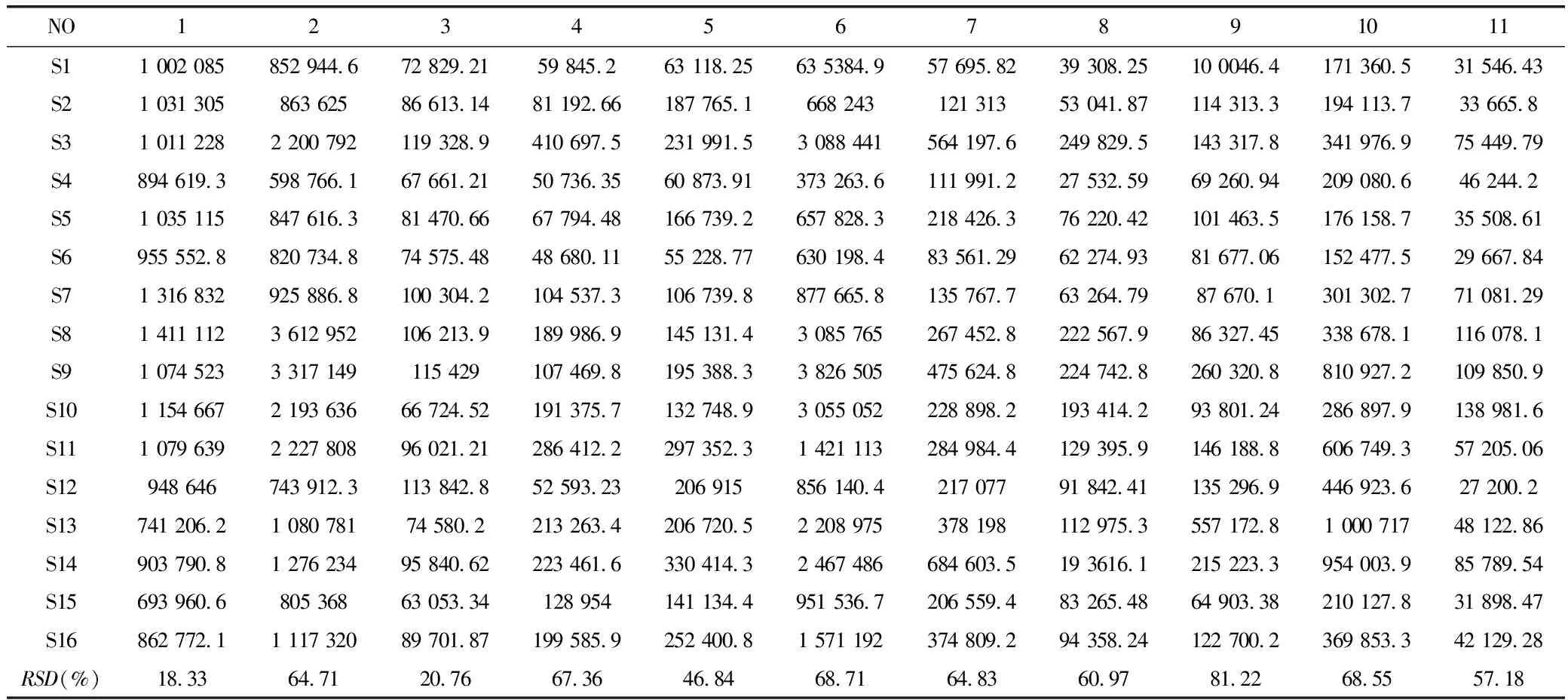
表5 马鞭草共有峰的峰面积
2.4.5 特征指纹图谱的结果分析 应用SPSS 23.0分析软件对16批马鞭草进行主成分分析(表6、表7、图3),以特征值>1为标准,选取前3个作为主成分,其方差累积贡献率达到84.233%。第一主成分特征值及方差贡献率均大于第二主成分,说明第一主成分包含信息最多。由旋转前后因子载荷矩阵可知,1号色谱峰、2号色谱峰和6号色谱峰在第一成分中有明显的正相负荷,表明戟叶马鞭草苷、马鞭草苷、毛蕊花苷可作为马鞭草的指标性成分,反映样品特性。
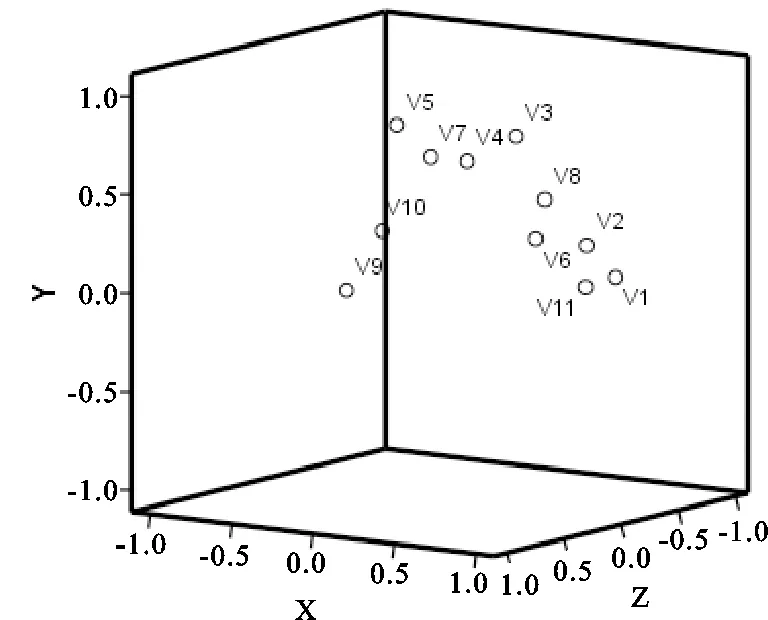
图3 样品主成分三维得分

表6 旋转前后的特征值及方差贡献率
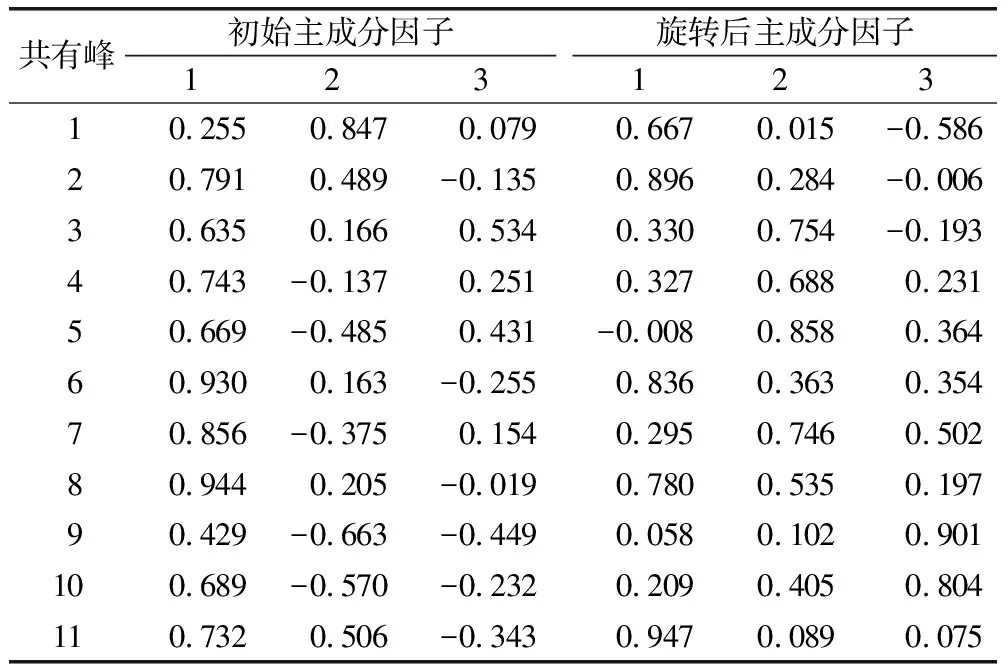
表7 因子载荷矩阵
将16批马鞭草样品特征图谱中11个共有峰的峰面积经均一化数据处理后作为变量,应用SPSS 23.0统计分析软件,采用中位数聚类法,区间选取平方Euclidean距离,进行聚类分析(图4)。当类间距为10时,16批药材可分为两大类,S3、S8、S9和S10分为一类,其余分为一类,与相似度评价结果较为一致。
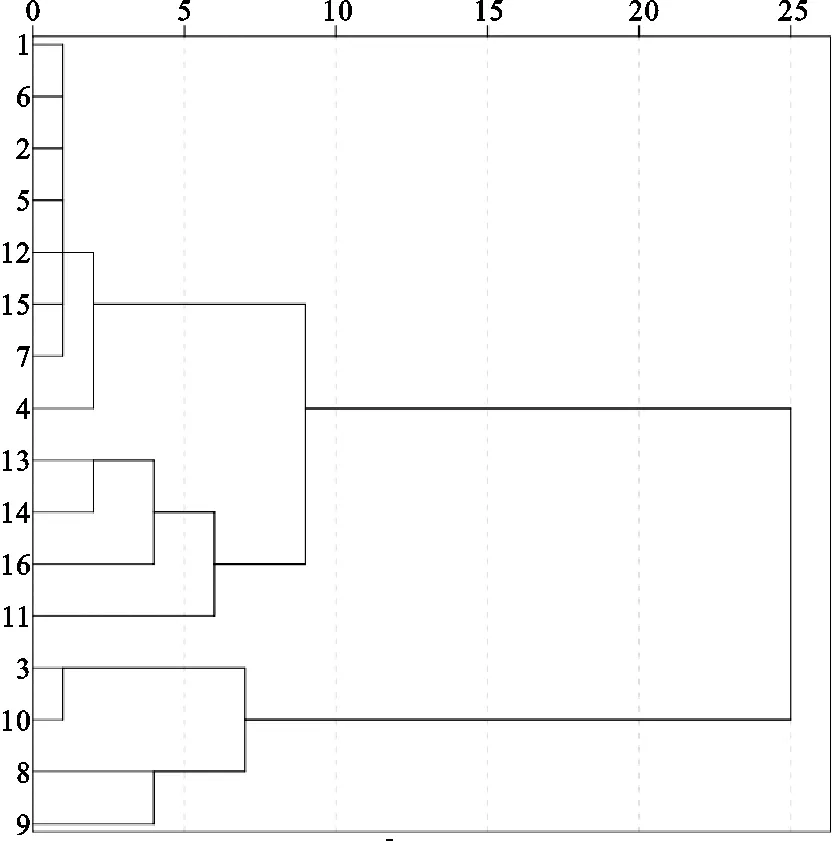
图4 聚类分析结果
2.5 指标性成分含量测定
2.5.1 线性关系考察 配制一系列含有戟叶马鞭草苷、马鞭草苷和毛蕊花糖苷的混合对照品溶液,按“2.3”项下条件进样测定,以进样浓度和峰面积分别为横、纵坐标,绘制标准曲线,得标准曲线:Y=1.0×106X+925.81,r=0.999 8;Y=1.0×106X+18 654,r=0.999 6;Y=956 465X+28 464,r=0.999 5。结果表明,戟叶马鞭草苷在0.143~3.580μg、马鞭草苷在0.232~5.810μg、毛蕊花糖苷在0.309~7.735μg范围内的进样量,与峰面积呈现良好的线性关系。
2.5.2 方法学考察 精密度试验。取“2.2”项下供试品溶液,按“2.3”项下色谱条件测定,连续进样6次,测得峰面积,并计算戟叶马鞭草苷、马鞭草苷和毛蕊花糖苷的含量和RSD值,3种成分的RSD值分别为1.03%、1.23%和1.16%,表明所用方法的精密度良好。
稳定性试验。取“2.2”项下供试品溶液,分别于0、2、4、6、8、10、12h检测,测得峰面积,并计算戟叶马鞭草苷、马鞭草苷和毛蕊花糖苷的含量和RSD值,供试品溶液中3种成分的RSD值分别为1.00%、0.63%和1.11%,表明所用方法的稳定性良好。
重复性试验。取药材(S3)粉末约0.5g,精密称定,按“2.2”项下方法制备供试品溶液6份,并按“2.3”项下色谱条件测定峰面积,计算戟叶马鞭草苷、马鞭草苷和毛蕊花糖苷的含量和RSD值,供试品溶液中3种成分的RSD值分别为1.70%、1.12%和1.71%,表明所用方法的重复性良好。
加样回收率试验。称取同一药材(S11)粉末6份,每份约0.1g,精密称定,置具塞锥形瓶中,分别加入戟叶马鞭草苷(0.628mg/mL)、马鞭草苷(0.828mg/mL)及毛蕊花糖苷(0.518mg/mL)3种对照品溶液1mL,按“2.3”项下色谱条件测定峰面积,计算样品中戟叶马鞭草苷、马鞭草苷和毛蕊花糖苷的含量和测定量,3种指标性成分平均回收率分别为97.83%、98.67%和97.76%,其RSD值分别为0.89%、0.87%和1.18%,见表8。

表8 3种指标性成分的加样回收率 (n=6)
2.5.3 含量测定 取药材粉末0.5g,精密称定,依照“2.2”项下方法制备供试品溶液并测定,按外标法计算样品中各成分的含量,结果见表9。
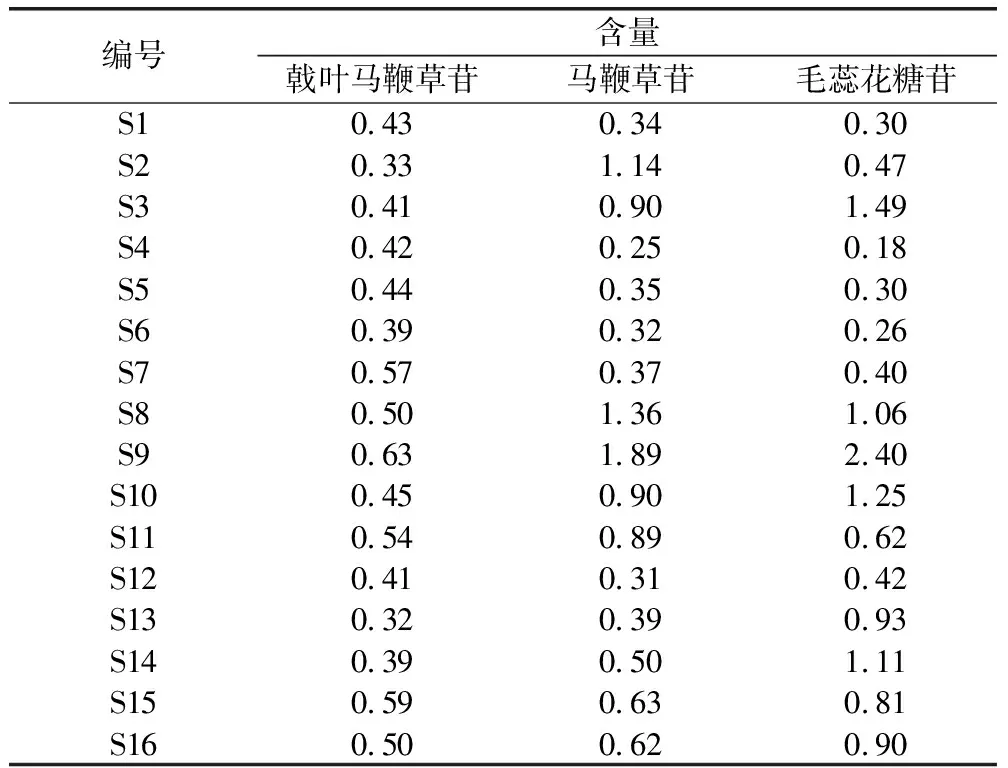
表9 指标性成分含量测定结果 (%)
不同批次马鞭草药材中戟叶马鞭草苷含量在0.32%~0.63%,马鞭草苷含量在0.25%~1.89%,毛蕊花糖苷含量在0.26%~2.40%。研究结果表明,不同产地不同批次马鞭草药材中各成分含量差异明显。
2.6 一测多评法测定药材中3种指标性成分的含量
2.6.1 相对校正因子的计算 在线性范围内,各成分的量与峰面积成正比关系,即W=fA,相对校正因子f(k/m)=fk/fm=(Wm×Ak)/(Wk×Am),其中Ak为内参物峰面积,Wk为内参物的质量或浓度,Am为组分m的峰面积,Wm为组分m的质量或浓度。以马鞭草苷为内参物,测定结果计算得f马鞭草苷/戟叶马鞭草苷=1.11,f马鞭草苷/毛蕊花糖苷=1.46,RSD分别为0.88%、2.68%,见表10。
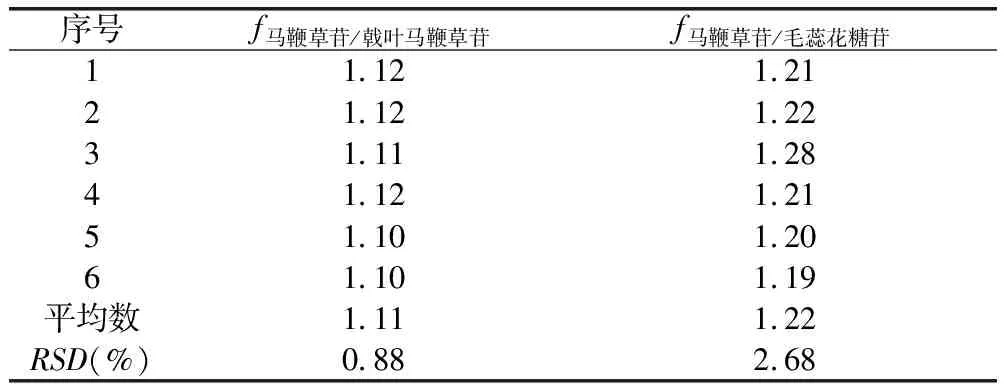
表10 3种成分的相对校正因子
2.6.2 相对校正因子重复性考察 精密吸取混合对照品溶液2、4、10、20、50μL,在确定的色谱条件下进样测定,计算得f马鞭草苷/戟叶马鞭草苷=1.15,f马鞭草苷/毛蕊花糖苷=1.44,RSD分别为2.69%、1.10%,结果显示,各成分的f值重复性良好。
2.6.3 不同色谱仪和色谱柱考察 取对照品溶液,选择Shimadzu LC-10A型和Agilent 1200型高效液相色谱仪,选定色谱柱Agilent C18(250mm×4.6mm,5μm)、Kromasil C18(250mm×4.6mm,5μm)进行色谱分析,计算f值。结果表明,不同高效液相和色谱柱对马鞭草3种指标性成分f值的影响无明显差异,见表11。
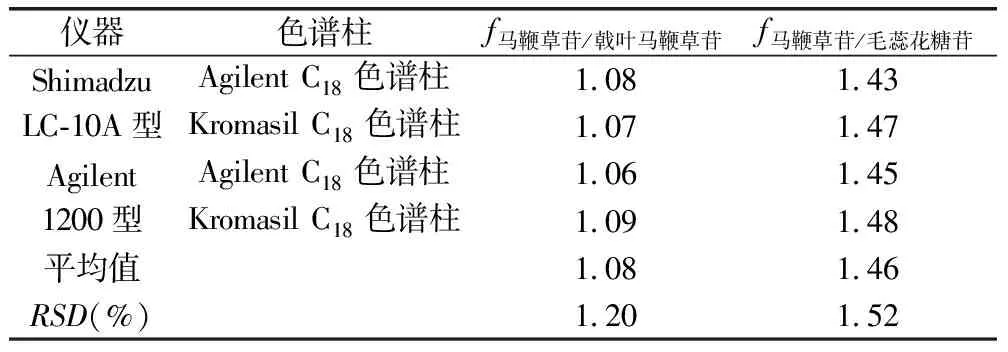
表11 仪器和色谱柱对相对校正因子的影响
2.6.4 不同流速考察 同等色谱条件下,分别设定流速为0.8、0.9、1.0mL/min,取混合对照品溶液进样测定,计算f值,RSD分别为0.53%、0.40%。表明流速对各成分f值的影响无明显差异。
2.6.5 不同柱温考察 在同等色谱条件下,分别设定柱温为25、30、35℃,取混合对照品溶液进样测定并计算f值,结果RSD为0.92%、0.40%。表明柱温对各成分f值的影响无明显差异。
2.6.6 待测成分色谱峰定位标准 以马鞭草苷为内参物,定位标准设置为保留时间差和相对保留值。由表10、表11 结果可知,保留时间差波动较大,RSD>2%。相对保留值波动较小,RSD均<2%,无明显差异。因此,选择相对保留值作为最终定位标准,见表12。

表12 相对保留值考察结果
2.7 一测多评法与外标法的比较
取药材粉末0.5g,制备供试品溶液进行测定,分别采用外标法和一测多评法计算各成分的含量。
由实验结果可知,戟叶马鞭草苷、毛蕊花糖苷对马鞭草苷的保留时间相对校正因子平均值分别为0.75、0.80。因此戟叶马鞭草苷保留时间T1=0.75×ts,毛蕊花糖苷保留时间T2=0.80×ts,其中ts为马鞭草苷的保留时间。戟叶马鞭草苷、毛蕊花糖苷对马鞭草苷的相对校正因子为1.11和1.22。根据研究结果得出含量计算公式:
戟叶马鞭草苷:lgCi=1.11×(lgCs×lgAi)/lgAs
毛蕊花糖苷:lgCi=1.22×(lgCs×lgAii)/lgAs
其中As为内标物马鞭草苷的峰面积,Cs为内标物马鞭草苷的浓度,Ai和Aii分别为戟叶马鞭草苷、毛蕊花糖苷的实测峰面积。测定结果见表13。

表13 外标法和一测多评法测定3种指标性成分的含量 (%)
3 讨论
建立马鞭草指纹图谱与主成分分析、聚类分析相结合的方法,可合理评价该药材质量,为建立科学的马鞭草药材质量控制方法提供依据[10]。
一测多评法可作为中药饮片、配方颗粒、中成药等中药制剂质量控制的有效手段[11]。本研究首次将一测多评法应用于马鞭草药材的质量评价体系中,具有快速、简便、低成本等优点,可有效应对大量分离纯化难度较高的化学对照品稀缺以及价格昂贵的局面,并提高检测效率,对马鞭草药材的评价标准的完善具有重大意义,也将进一步推动一测多评法在药材质量控制中的应用。
本研究通过建立马鞭草指纹图谱,同时运用一测多评法对16批马鞭草中3种指标性成分进行含量测定,不同批次间戟叶马鞭草苷、马鞭草苷及毛蕊花糖苷的含量差异不大。两种方法测定结果无明显差异,相对校正因子重复性良好。通过马鞭草苷可实现对其余两种指标性成分的定量分析,方法简单准确,并且能节省大量对照品,经济实用,具有一定的推广价值。
