反向离子对高效液相色谱法测定地夸磷索钠含量
2023-06-11张小平朱永星
张小平,魏 渊,朱永星
(1. 江苏大学药学院,江苏 镇江 212013; 2. 江苏恒新药业有限公司,江苏 镇江 212009)
干眼症的发病原因包括泪液质和量异常,动力学异常的内膜稳定性下降,眼部神经异常,眼部炎症导致角膜、结膜细胞损伤等,我国发病率为21%~30%[1],且趋于年轻化、大众化[2-3]。目前,治疗干眼症的常规方法是运用人工泪液或联合抗炎药物滴眼[4],常用药物有聚乙二醇[5]、普拉洛芬联合玻璃酸钠[6]、石斛夜光丸联合玻璃酸钠[7]、环孢素A[8]等。3%地夸磷索钠滴眼液于2010 年在日本上市,2017 年10 月进入中国市场,临床治疗干眼症疗效良好[9-13]。地夸磷索钠作用于眼结膜上皮及杯状细胞膜上的P2Y2 受体,通过上调细胞内的钙离子浓度,促进水分及黏蛋白的分泌,进而改善干眼症症状,是一种治疗干眼症的新型药物[14-17]。截至2022 年5 月25 日,国内尚无药品生产企业取得地夸磷索钠滴眼液批准文号,也无其含量测定的相关报道。本研究中采用反向离子对高效液相色谱法测定了地夸磷索钠的含量[18]。现报道如下。
1 仪器与试药
1.1 仪器
BP211D 型电子天平(德国赛多利斯公司,精度为十万分之一);UV - 2600i 型紫外可见分光光度计,LC - 2030 Plus 型高效液相色谱仪,LC - 20 AD 型高效液相色谱仪,均购自日本岛津公司;Agilent 1220 infinity LC 型高效液相色谱仪(美国安捷伦科技公司);DL -204 型电热恒温干燥箱(天津市中环实验电炉有限公司);KQ-800DE型数控超声波清洗器(昆山市超声仪器有限公司,功率为1 600 W,频率为40 kHz)。
1.2 试药
地夸磷索钠对照品(广东先强药业有限公司,批号为211002,纯度为99.8%);地夸磷索钠(批号分别为220301,220302,220303),纯化水,均由江苏恒新药业有限公司自制;乙腈(德国默克集团);磷酸二氢钾、氢氧化钾(国药集团化学试剂有限公司);四丁基硫酸氢铵(上海麦克林生化科技股份有限公司)。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Thermo Scientific™C18柱(100 mm×4.6 mm,3µm);流动相:磷酸盐缓冲液(pH 6.8)-乙腈(85∶15,V/V);流速:1.0 mL/ min;检测波长:262 nm;柱温:30 ℃;进样量:10µL。
2.2 溶液制备
取地夸磷索钠样品10 mg,精密称定,置100 mL 容量瓶中,加纯化水适量,超声(功率为1 600 W,频率为40 kHz)20 min 使其彻底溶解,加纯化水定容,摇匀,即得供试品溶液。取地夸磷索钠对照品适量,置电热恒温干燥箱中干燥至恒重,取10 mg,精密称定,置100 mL容量瓶中,加纯化水适量,超声使溶解,加纯化水稀释并定容,即得对照品溶液。
2.3 方法学考察
专属性试验:分别取2.2 项下对照品溶液、供试品溶液,以纯化水为阴性对照品溶液,各10µL,按2.1 项下色谱条件进样测定,结果对照品溶液在此色谱条件下的理论板数按地夸磷酸钠峰计为6 237,拖尾因子为0.909。地夸磷索钠理论板数不低于4 000,拖尾因子应不大于1.5,按外标法以峰面积计算。供试品溶液色谱中,在与对照品溶液色谱相同保留时间处有相应色谱峰。色谱图见图1。
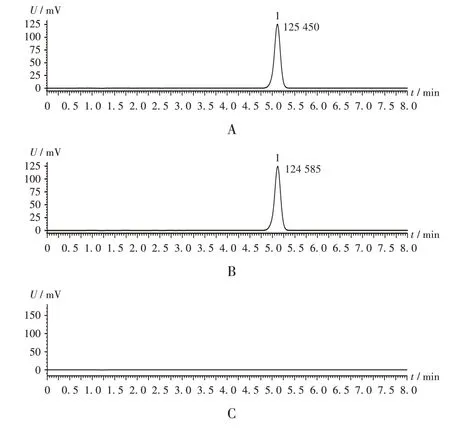
1. 地夸磷索钠A. 对照品溶液 B. 供试品溶液 C. 阴性对照品溶液图1 高效液相色谱图1.Diauafosol sodiumA.Reference solution B.Test solution C.Negative reference solutionFig.1 HPLC chromatograms
破坏性试验:取地夸磷索钠样品100 mg,精密称定,置100 mL 容量瓶中,按2.2 项下方法制备浓度为1 mg/mL的供试品溶液,平行6份,除1份溶液未经破坏外,其余溶液分别经强酸(2 mol/L HCl溶液、室温3 h)、强碱(2 mol/L NaOH溶液、室温3 h)、强氧化(3%H2O2溶液室温3 h)、热(100 ℃水溶0.5 h)、强光[(4 500±500)Lx照射1 d]条件破坏,破坏后,调pH至中性,加纯化水衡释为质量浓度为100µg/mL的溶液,按2.1项下色谱条件进样测定,结果供试品溶液经酸破坏后峰面积减小,热破坏后峰面积增大,对碱、氧化、强光破坏相对稳定,各降解产物均能较好分离,且对地夸磷索钠主峰无干扰。
检测限与定量限确定:取2.2 项下对照品溶液适量,加纯化水逐级稀释,按2.1项下色谱条件进样测定,分别以信噪比(S/N)为3和10时待测成分的质量浓度作为检测限和定量限。结果地夸磷索钠检测限为0.01µg/mL,定量限为0.03µg/mL。
线性关系考察:取地夸磷索钠对照品0.032 36 g,精密称定,置50 mL容量瓶中,加纯化水适量,逐级稀释成质量浓度分别为3.23,32.30,161.50,323.00,484.50,646.00µg/mL 的系列溶液,按2.1项下色谱条件进样测定。以地夸磷索钠质量浓度(X,µg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程Y=11 346X+35 924(r=0.999 9,n=6)。结果表明,地夸磷索钠质量浓度在3.23~646.00 µg/ mL 范围内与峰面积线性关系良好。
精密度试验:取地夸磷索钠对照品适量,加纯化水分别制成质量浓度为40,100,400µg/mL 的溶液,在日内各重复进样5 次,并连续测定5 d。结果地夸磷索钠低、中、高质量浓度的对照品溶液日内及内间精密度试验结果的RSD均≤2.0%(n= 5)。取同一批(批号为220301)样品适量,按2.2 项下方法配制成质量浓度为100µg/mL 的供试品溶液,在同一试验室的不同时间,由不同操作人员分别用岛津LC - 2030 Plus 型、Agilent 1220 infinity LC 型、岛津LC-20 AD 型高效液相色谱仪测定样品含量,结果中间精密度试验结果的RSD为0.52%。上述结果表明仪器精密度良好。
重复性试验:取样品(批号为220301)适量,按2.2项下方法配制成质量浓度为100 µg/ mL 的供试品溶液,平行5 份,按2.1 项下色谱条件各进样测定2 次,记录色谱图。结果地夸磷索钠的平均含量为99.60%,RSD为0.36%(n=10),表明方法重复性良好。
稳定性试验:取样品(批号为220301)0.010 05 g,精密称定,按2.2项下方法配制成质量浓度为100µg/mL的供试品溶液,分别于室温放置0,1,2,4,6,8,10,12 h时按2.1 项下色谱条件进样测定,记录色谱图。结果的RSD为0.69%(n= 8),表明供试品溶液在室温下放置12 h内稳定性良好。
加样回收试验:取已知含量的样品(批号为220301)适量,共9份,分别置100 mL容量瓶中,分别加入质量浓度为0.401 0 mg / mL 的对照品溶液20,25,30 mL,各3 份,如纯化水适量,超声使溶解,加纯化水定容,按2.1项下色谱条件进样测定,记录色谱图。结果见表1。
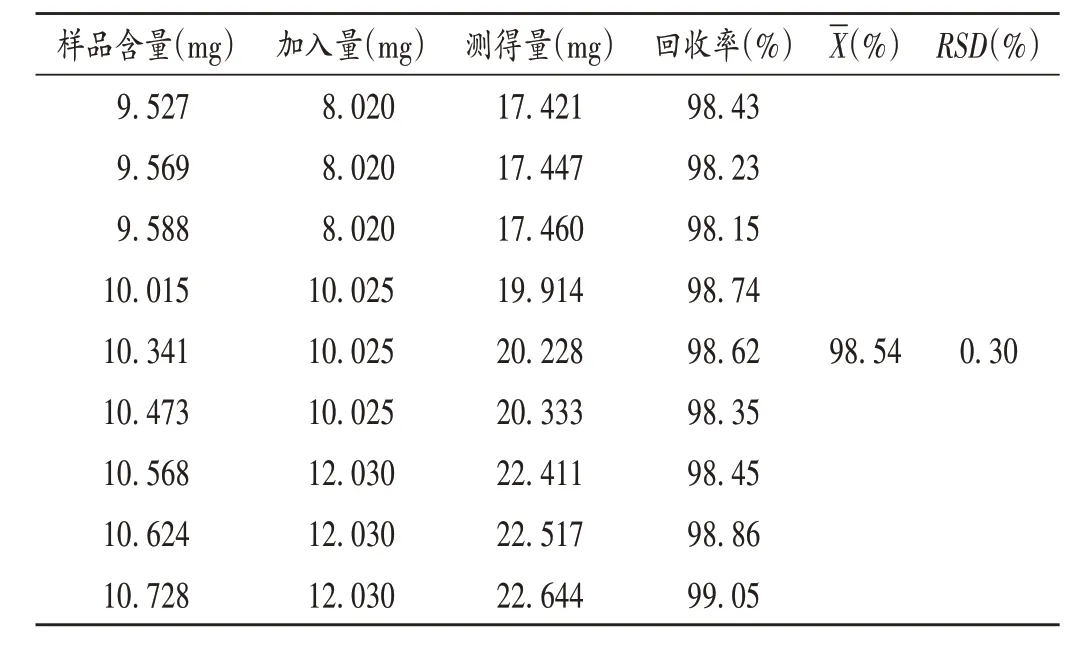
表1 地夸磷索钠加样回收试验结果(n=9)Tab.1 Results of the recovery test of diauafosol sodium(n=9)
2.4 样品含量测定
取3 批(批号分别为220301,220302,220303)样品各适量,按2.2 项下方法制备供试品溶液,按2.1 项下色谱条件分别进样测定2次,记录峰面积,并计算含量。结果平均含量分别为99.84%,99.97%,99.35%,RSD分别为0.13%,0.06%,0.20%(n=2)。
3 讨论
3.1 检测波长选择
前期试验中,取对照品10 mg,精密称定,加纯化水,制成每1 mL 溶液中含地夸磷索钠50µg 的溶液,采用紫外分光光度计于200~400 nm 波长范围内进行扫描。结果地夸磷索钠溶液约在205 nm 及262 nm 波长处有最大吸收,由于205 nm 只检测到该峰末端部分,属紫外末端吸收,故检测波长选择262 nm。
3.2 色谱柱选择
C18或C8反相色谱柱常用于分离弱极性、中性、非极性化合物,用C18或C8短柱(100 mm×4.6 mm,3µm)分离地夸磷索钠,保留时间为1.0 min,换为长柱(250 mm×4.6 mm,5µm)保留时间仅延长至2.0 min,由于地夸磷索钠显极性,故在反相色谱柱中的保留时间极短。
3.3 磷酸盐缓冲液配制方法选择
地夸磷索钠解离常数为6.3,在流动相为pH 6.8的磷酸盐缓冲液中以解离型居多,若选择氢氧化钠调节pH,溶液中的钠离子会对药物中的钠离子形成干扰,降低含量测定的准确度,偏差变大;若将pH 调节剂换为氢氧化钾,可避免外源性钠离子对测定结果的干扰。地夸磷索钠为弱酸类物质,流动相中需加入季铵类离子对(四丁基硫酸氢铵)[19]等正离子与药物磷酸基团的负离子(反离子)生成不带电荷的中性离子对,增加供试品在反相色谱柱中的溶解度,增大分配系数,改善分离效果,延长保留时间,实现样品充分分离。故磷酸盐缓冲液配制方法为取磷酸二氢钾6.80 g,氢氧化钾1.316g,四丁基硫酸氢铵5.0 g,加纯化水1 000 mL,调pH 至6.8。同时发现,四丁基硫酸氢铵缓冲盐溶液室温放置8 h 以上可见浑浊,故需临用现配。
3.4 流动相比例选择
随着流动相中乙腈浓度的增大,药物出峰时间提前,保留时间变短,为了延长药物在色谱柱中的保留时间,建议流动相中乙腈浓度不超过20%,故选择磷酸盐缓冲液与乙腈体积比为85∶15(V/V)。
3.5 进样时间选择
样品连续进样时间不宜过长,超过6 h,色谱峰逐渐漂移,峰形走样变形,理论板数变小,拖尾因子变小;超过10 h,单峰变为双峰,理论板数进一步变小,柱效下降,易导致泵和色谱柱损坏。故建议进样时间控制在4 h内,试验结束后需加倍延长冲洗色谱柱的时间。
3.6 方法评价
本研究中建立的方法专属性强、结果准确,可为地夸磷索钠原料药及其制剂的研发提供质量评价依据,为其正式标准的拟订提供参考。
