Current and novel modalities for management of chronic hepatitis B infection
2023-05-30ImanIbrahimSalamaSamiaSamiSomaiaSalamaGhadaAbdelLatifFatmaShaabanWalaaFouadAidaAbdelmohsenHalaRaslan
Iman Ibrahim Salama, Samia M Sami, Somaia I Salama, Ghada A Abdel-Latif, Fatma A Shaaban, Walaa A Fouad, Aida M Abdelmohsen, Hala M Raslan
Iman Ibrahim Salama, Somaia I Salama, Ghada A Abdel-Latif, Walaa A Fouad, Aida M Abdelmohsen, Department of Community Medicine Research, National Research Centre, Giza 12411, Dokki, Egypt
Samia M Sami, Fatma A Shaaban, Department of Child Health, National Research Centre, Giza 12411, Dokki, Egypt
Hala M Raslan, Department of Internal Medicine, National Research Centre, Giza 12411, Dokki,Egypt
Abstract Over 296 million people are estimated to have chronic hepatitis B viral infection(CHB), and it poses unique challenges for elimination. CHB is the result of hepatitis B virus (HBV)-specific immune tolerance and the presence of covalently closed circular DNA as mini chromosome inside the nucleus and the integrated HBV. Serum hepatitis B core-related antigen is the best surrogate marker for intrahepatic covalently closed circular DNA. Functional HBV “cure” is the durable loss of hepatitis B surface antigen (HBsAg), with or without HBsAg seroconversion and undetectable serum HBV DNA after completing a course of treatment. The currently approved therapies are nucleos(t)ide analogues,interferon-alpha, and pegylated-interferon. With these therapies, functional cure can be achieved in < 10% of CHB patients. Any variation to HBV or the host immune system that disrupts the interaction between them can lead to reactivation of HBV. Novel therapies may allow efficient control of CHB. They include direct acting antivirals and immunomodulators. Reduction of the viral antigen load is a crucial factor for success of immune-based therapies. Immunomodulatory therapy may lead to modulation of the host immune system. It may enhance/restore innate immunity against HBV (as toll-like-receptors and cytosolic retinoic acid inducible gene I agonist). Others may induce adaptive immunity as checkpoint inhibitors, therapeutic HBV vaccines including protein(HBsAg/preS and hepatitis B core antigen), monoclonal or bispecific antibodies and genetically engineered T cells to generate chimeric antigen receptor-T or Tcell receptor-T cells and HBV-specific T cells to restore T cell function to efficiently clear HBV. Combined therapy may successfully overcome immune tolerance and lead to HBV control and cure. Immunotherapeutic approaches carry the risk of overshooting immune responses causing uncontrolled liver damage. The safety of any new curative therapies should be measured in relation to the excellent safety of currently approved nucleos(t)ide analogues.Development of novel antiviral and immune modulatory therapies should be associated with new diagnostic assays used to evaluate the effectiveness or to predict response.
Key Words: Current modalities; Novel modalities; Management; Chronic hepatitis B infection; Direct acting antiviral therapy; Immunotherapy; Therapeutic vaccination
INTRODUCTION
Hepatitis B is a potentially life-threatening liver infection caused by the hepatitis B virus (HBV). It is a major global health problem. It can cause chronic infection and puts people at high risk of death from cirrhosis and liver cancer[1]. In 2019, the World Health Organization (WHO) reported that there were three million people living with new HBV infection and 296 million people living with chronic HBV(CHB) infection. The target of the Sustainable Development Goals and Global Health Sector Strategy in 2020 to decrease the incidence of hepatitis B has been met, as the global prevalence of CHB infection among children aged < 5 years decreased to less than 1% (0.94%) in 2019 compared to 5% in the prevaccine era[2]. The prevalence of CHB ranges from < 2% in the United States and Western Europe to ≥8% in western Africa[3]. The endemicity of HBV in Asia is heterogeneous, and most of the region has a moderate to high prevalence of HBV infection, except for a few low endemic areas.
The WHO[4] reported that in 2019 approximately 820000 deaths were related to HBV infection,mainly from cirrhosis and hepatocellular carcinoma (HCC). A National Egyptian study reported that among vaccinated children aged 1-16 years, 1535 (42.8%) were identified to have non-seroprotective levels of anti HBs (< 10 IU/L), and CHB prevalence was 0.39%[5,6]. Nearly, 95% of infections occurring during infancy and early childhood progress to CHB[7]. Over 90% of infections occurring during adulthood resolve as a result of developing robust immune responses[8]. In naïve CHB patients, the cumulative incidence of hepatic cirrhosis within 5 years of infection is about 10%-20% among patients with active CHB, and about 2%-5% develop liver cancer annually[9]. Infections during infancy usually carry a greater risk of developing cirrhosis and liver cancer[10].
HBV can be transmitted by sexual exposure, perinatal (mother-to-child transmission), percutaneous,and direct contact with the blood and other body fluids of HBV-infected persons[11]. Acute HBV infection manifestations range from subclinical to icteric to fulminant hepatitis in few cases. Patients who resolved the acute infection develop an efficient B cell immune response displayed by high levels of antibodies (anti-HBs) to surface antigen (HBsAg). They also develop a vigorous T cell response including CD4 and CD8 T cells to produce antiviral cytokines that kill infected hepatocytes. The difference between patients who resolved the acute HBV infection and those with CHB infection is primarily dependent on the magnitude and effectiveness of their immune response. CD8+ T cells are considered responsible for viral clearance during acute recovery but are impaired by continuous exposure to high viral HBsAg and immunosuppression in CHB[12]. The patient immune response directed towards the virus-infected hepatocytes usually leads to hepatocytes damage with an increasing risk of liver cirrhosis and HCC due to long-term liver inflammation associated with lack of viral clearance[13]. The present review summarized the current and novel diagnostic and therapeutic modalities for HBV infection.
HBV LIFE CYCLE
HBV is a DNA hepatotropic virus[14]. HBV is a double-stranded DNA virus that belongs to the Hepadnaviridae family. It consists of an outer envelope containing HBsAg and a capsid core. The capsid core bears the viral genome and DNA polymerase[15]. Inside the host nucleus, the relaxed circular DNA genome is transformed to a covalently closed circular DNA (cccDNA), which is the stable form of the virus acting as a mini-chromosome. The cccDNA produces a template for generating subgenomic mRNAs and pre-genomic RNA (pgRNA) and uses the host transcription factors for viral synthesis[16].HBV polymerase catalyzes the pgRNA to synthesize the viral genomic DNA, while the mRNA is translated to various viral proteins as a part of the HBV life cycle[17]. The HBV viral genome encodes RNA transcripts and seven proteins. The DNA polymerase gene is the main part of the virus genome sequence. The S gene includes three parts: pre-s1, pre-s2, and s encoding envelope HBsAg. The C gene encodes both hepatitis B core (HBcAg) and “e” antigens (HBeAg). The HBx gene is encoded in both the polymerase gene and the X region[18,19].
There are three different sized proteins derived from the envelope gene: Small from s (SHB), middle from pre-s2 + s (MHB) , and large from pre-s1 + pre-s2 + s (LHB)[20]. The virus secretes many defective particles as enveloped nucleocapsids that are empty or contain defective immature genomes and subviral lipid particles (SVPs) containing the viral surface antigens. These SVPs are noninfectious smaller particles containing the most plentiful form of 17-25 nm spherical particles of SHB. They also contain filamentous (or tubular) particles of variable length and comprised of SHB, MHB, and LHB proteins[21]. HBsAg proteins can gather to noninfectious SVPs approximately 22 nm in diameter containing the HBV envelope proteins (LHB, MHB, and SHB) and are secreted at a level 1000-fold to 100000-fold higher than the infectious particles[22].
During the chronic phase, HBsAg may lead to dysregulation of innate and adaptive host immunity through interacting with either the immune or non-immune cells causing impairment of the immune system and liver damage. SVPs inhibit antibody responses to the virus[23]. HBsAg could activate the unfolded protein response leading to cellular premalignant changes,i.e.it is associated with both hepatic inflammation and HCC[24]. LHB plays a crucial role in the virus envelope and initiates infectivity[25], and the form of the HBV particles appears to be determined by the ratio of the various HBsAg proteins[26]. SHB may have a role in HCC metastasis and progression. Among CHB patients,SHB can be a target for prevention or intervention of HCC progression. A continuous low level of HBsAg in CHB patients is due to failure of the host to eliminate it due to several effects related to the virus, host, and other factors[27]. Moreover, HBeAg and HBx are also major carcinogenesis-related proteins as they are essential to initiate and maintain virus replication. Various cellular events are related to HBx, such as ubiquitination, autophagy, epigenetic modifications, and non-coding RNA regulation. Subsequently, they might contribute to hepatic inflammation, fibrosis, and HCC[28].
After resolution of an acute infection, there is disappearance of serum HBV DNA, appearance of anti-HBc, HBeAg seroconversion to anti-HBe, and finally HBsAg seroconversion to anti-HBs during recovery[11]. Occult HBV infection is defined by the presence of isolated anti-HBc with the absence of HBsAg and anti-HBs[29,30]. HBV DNA is a direct measurement of the viral load. It indicates viral replication activity and disappears at the recovery from HBV infection or gradually diminishes in CHB.High titers of HBV DNA may lead to more quickly to liver cirrhosis and HCC[31]. HBV DNA is immunostimulatory and is recognized by cyclic GMP-AMP synthase and stimulator of interferon genes.Lauterbach-Rivièreet al[32] found that in infected hepatocytes HBV passively escapes recognition by cellular sensors of nucleic acids by producing non-immunostimulatory RNAs and avoiding sensing of its DNAs by cyclic GMP-AMP synthase/stimulator of interferon genes without active inhibition of the pathway.
NEW HBV BIOMARKERS
Novel biomarkers such as hepatitis B core-related antigen (HBcrAg) and serum HBV RNA are recognized as important markers to monitor the antiviral effects of the emergent therapies. HBcrAg is made up of three related viral proteins sharing an identical 149 amino acid sequence. These include HBcAg, HBeAg, and a truncated 22 kDa pre-core protein[33]. Several Asian and European studies reported a significant positive correlation between serum HBcrAg levels and the amount of intrahepatic cccDNA[34]. The presence of HBcrAg among patients with undetectable HBV DNA indicates continued transcription of cccDNA and can predict clinical relapse and be an aid for clinicians in identifying patients with a higher risk of HCC development[35,36]. HBcrAg can monitor the response to novel therapy targeting cccDNA[37]. The combination of both HBsAg and HBcrAg was found to be an outstanding biomarker for assessing the risk of HCC[38]. Treated patients with persistently high HBcrAg levels had a higher risk of HCC[39]. In addition, HBcrAg is a good virologic marker differentiating active HBV from inactive HBV in the presence of negative HBeAg[40].
The level of serum HBV RNA, presented as a pgRNA-containing virion, is correlated with intrahepatic pgRNA and cccDNA content, and it signifies high viral replication activities[39,41]. pgRNA is present in serum at lower levels than HBV DNA in treatment-naïve patients. It is enriched during nucleos(t)ide analogue (NA) therapy, which inhibits reverse transcriptase activities by blocking the transcription of pgRNA into HBV DNA. This could explain the presence of HBV RNA in serum after NA therapy, despite the undetectable serum HBV DNA[39,42]. The HBV RNA level was found to be associated with a higher risk of HCC and recurrence in patients treated with NAs[43,44]. However, the way for serum HBV RNA detection and assessment should be standardized before its clinical application.
Yanget al[45] found that HBV can encode HBV-related microRNA and named it HBV microRNA 3(HBV-miR-3). It is used in the monitoring of HBV infection, and it is positively correlated to HBV DNA,HBsAg, and pgRNA. HBV-miR-3 was secreted by HBV-infected hepatocytes and existed in the peripheral blood exosomes of CHB patients. It is positioned at site 373–393 in the HBV genome and can be encoded by three mRNAs, (except for HBx mRNA). HBV-miR-3 and pgRNA are synthesized using cccDNA. Little effect on HBsAg, pgRNA, and HBV-miR-3 was found after antiviral drug treatment[46].
MECHANISMS OF HBV REACTIVATION
The integrated HBV DNA in the host genome can produce viral RNAs and proteins, although it does not produce progeny virus[47]. In the early HBV life cycle, HBV DNA integration occurs at the double stranded breaks throughout the whole host genome. Mutants of HBV genes may be the products of HBV DNA integration, which can stably express mutant HBV proteins. This may lead to HCC and might be a useful biomarker to monitor disease progression from CHB to HCC[48]. HBV reactivation may occur among HBV patients with resolved infection or inactive carrier state and spontaneously or as a complication of immunosuppressive therapy[7]. The basic initial step is the loss of HBV immune control. The Asian Pacific Association for the Study of the Liver (APASL) recommends the need to screen all patients for hepatitis B preceding the initiation of immunosuppressive therapy and to administer protective NAs to those patients with a considerable risk of hepatitis and acute on chronic liver failure due to hepatitis B reactivation[49].
CURRENT THERAPIES FOR CHB
The current available treatments can control viral replication and decrease HCC progression, but these regimens are not curative and may require lifelong therapy[50].
Interferon-alpha and pegylated-interferon-alpha
Interferon-alpha (IFN-α) and pegylated-IFN-α (PEG-IFN-α) act as immune modulators of finite duration.They work through inducing immune-mediated control of HBV infection with long-lasting viral replication suppression after therapy[10,50]. They inhibit viral cell entry, increase host immune response, induce cccDNA degradation, inhibit pgRNA encapsidation, and exert epigenetic modification of cccDNA transcription. Epigenetically they suppress cccDNA transcription and viral protein secretion[51], with a higher success rate shown in PEG-IFN compared to IFN[52]. IFN-α and PEG-IFN-α are the only licensed anti-HBV therapies capable of eliminating cccDNA[53]. PEG-IFN-α-2a and PEG-IFN-α-2b improve the expression of innate antiviral genes and proteins, stimulate natural killer (NK) T and CD8 immune cells, and enhance a non-cytolytic viral clearance through cytokines or cytolysis of the infected cells[54].
They have high HBsAg clearance rates, especially with genotypes A and B. However, the rate of success of IFN therapy is still low with major side effects. The long-term 5-year HBsAg loss post treatment is less than 20%[55]. A study by Wuet al[27] demonstrated that after a short-course of PEGIFN-α-2b re-treatment in patients with HBsAg recurrence a high rate of functional cure could be achieved after therapy withdrawal, which was relatively safe.
Nucleo(t)ide therapy
NAs are nucleos(t)ide reverse transcriptase inhibitors. As an oral therapy targeting viral reverse transcriptase activity, it inhibits viral replication and prevents new HBV DNA formation from pgRNA[56]. However, it has minimal effects on the existing or newly formed cccDNA because cccDNA formation does not depend on the viral reverse transcriptase activity. This allows viral relapse after NA withdrawal[57]. Trials with finite treatment duration have been conducted[58]. There are currently seven approved NAs, mainly tenofovir disoproxil fumarate (TDF), tenofovir alafenamide, and entecavir.They have high effectiveness in decreasing the risk of cirrhosis and HCC with good tolerability and minimal side effects[10,11]. However, HBV disease progression can still occur even with sustained viral suppression[39]. The probability of HBsAg loss after NA cessation varies according to patient ethnicity,HBV genotype, and viral antigen levels at the end of treatment. Non-Asian patients are more likely to achieve favorable outcomes[59]. Irrespective of ethnicity, lower serum levels of HBcrAg and HBsAg and HBV genotype C are associated with higher rates of viral response, HBsAg loss, and lower rates of alanine aminotransferase (ALT) flares[60].
NAs inhibit viral replication but not viral transcription or translation[61]. The newly developed generation of NAs should be safer and more efficient through novel prodrug methods. ATI-2173 is a noncompetitive non-chain terminating clevudine derivative that can inhibit HBV polymerase[62]. After dosing for 28 d, the mean HBV DNA reduced to 2.8 Log10 IU/mL without any serious adverse events in phase I studies[63]. But during the short dosing interval, no changes were seen in HBsAg levels.Pradefovir, HS-10234, and NCO-48 fumarate are other new NA prodrugs derived from adefovir and tenofovir and increase antiviral potency and reduce metabolite toxicity[64,65]. Similar effectiveness is derived from TDF. Agents targeting the RNase H function of the HBV polymerase are in preclinical development[66].
When HBV DNA was undetectable, NAs had little effect on the HBV-miR-3 levels. PEG-IFN following NA therapy had a positive impact on the decrease of HBV-miR-3, pgRNA, and HBsAg[46].
Patient selection and therapy withdrawal
The three major liver societies [the American Association for the Study of Liver Diseases (AASLD), the European Association for the Study of the Liver, and APASL] strongly agree that patients with active disease (defined as those with elevated HBV DNA and ALT levels), those with decompensated liver disease, and those with cirrhosis should receive treatment[67]. Table 1 summarized the current therapy regimens for CHB as recommended by AASLD 2023[68].
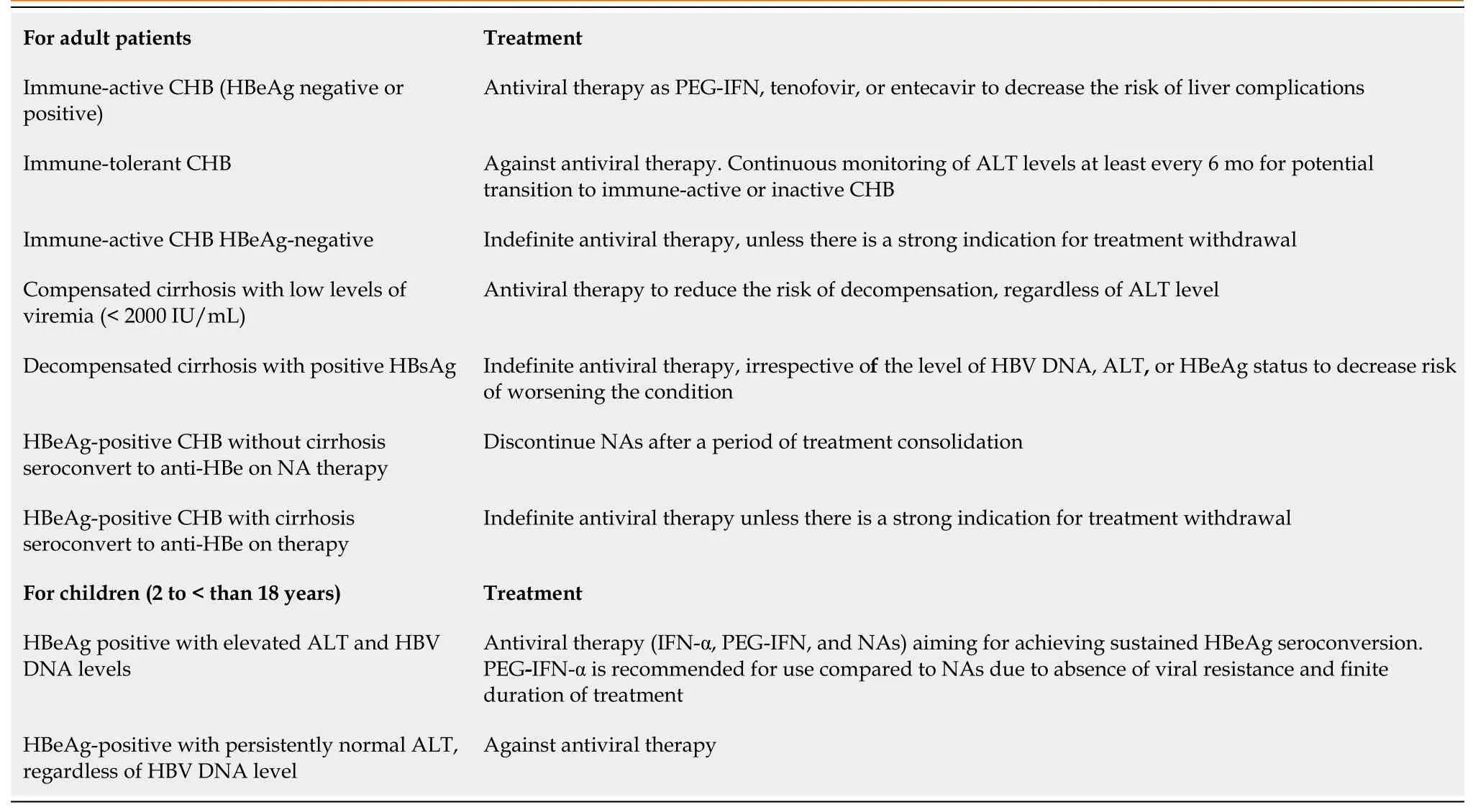
Table 1 Current therapy regimens for chronic hepatitis B viral infection as recommended by the American Association for the Study of Liver Diseases, 2023[68]
After seroconversion and consolidation therapy, drug withdrawal may be considered as recommended by guidelines in HBeAg-positive patients[69]. The European Association for the Study of the Liver guidelines in 2017[10] recommended a minimum of 3 years of viral inhibition, while the AASLD guidelines in 2018[70] recommended at least 2 years of combined therapy and viral inhibition.In 2021, APASL guidelines recommended NA withdrawal in HBeAg-positive patients who experienced HBeAg seroconversion and a 3-year consecutive treatment in HBeAg-negative patients who have had undetectable HBV DNA with normalization of ALT[71]. The disagreement of when antiviral therapy should be discontinued in CHB patients is due to the lack of effective methods for assessing cccDNA in the hepatocyte nucleus[72,73]. Moreover, stopping NAs carries a high incidence of virological relapse and surge of ALT levels, with high risk of fibrosis progression, decompensation, and HCC. Over 40% of CHB patients who stopped NAs eventually received re-treatment[71].
NEW TREATMENT MODALITIES
Complete cure is defined as an undetectable HBsAg and HBV DNA in the serum and complete eradication of both the intrahepatic cccDNA and integrated HBV DNA. Functional cure is defined as sustained undetectable levels of both HBsAg and HBV DNA in the serum with or without anti-HBs seroconversion and the persistence of low amounts of intrahepatic cccDNA and HBV DNA integration[74]. Since it is currently not possible to eradicate the cccDNA mini-chromosome or the integrated HBV DNA from the infected cell, functional cure is more attainable with the approved therapies. In addition,to reach high rates of HBsAg loss, there is a crucial need for short duration regimens because current treatments do not restore the immune dysfunction occurring in CHB.
Ongoing efforts are directed towards developing novel anti-HBV drugs that achieve a complete cure.Two main classes of novel therapies are recognized: (1) Agents that directly interfere with the HBV life cycle; and (2) Agents that reinforce the host immune response against HBV infection (immune modulators). The combination of these two novel agents is expected to be more effective. Antiviral immune responses should be restored to allow elimination of both cccDNA and integrated HBV.Figure 1 shows the HBV life cycle and the effects of current and novel therapies. Table 2 shows the new HBV treatment modalities and its clinical trial phases.
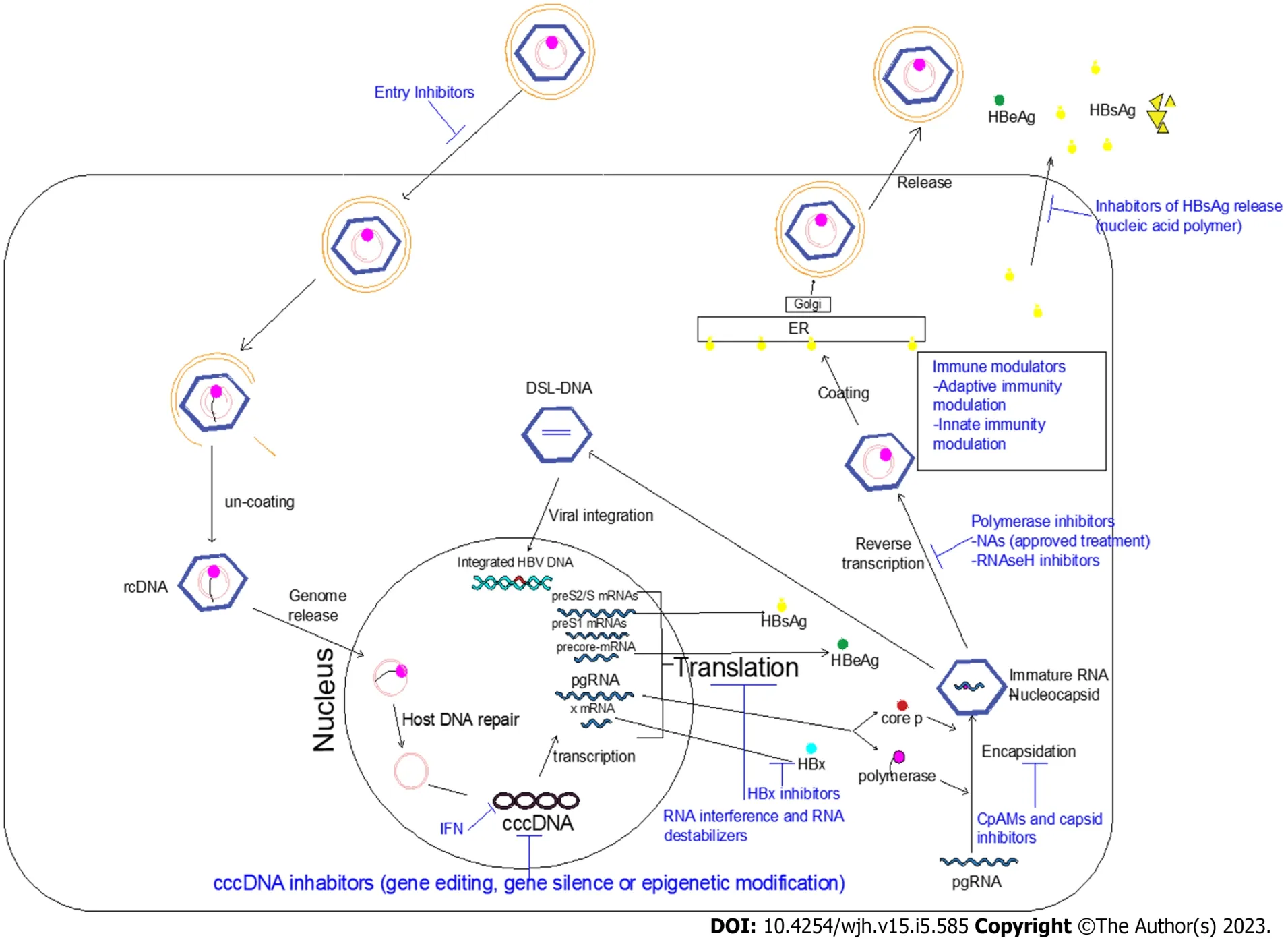
Figure 1 Hepatitis B virus life cycle and the effects of current and novel therapies.
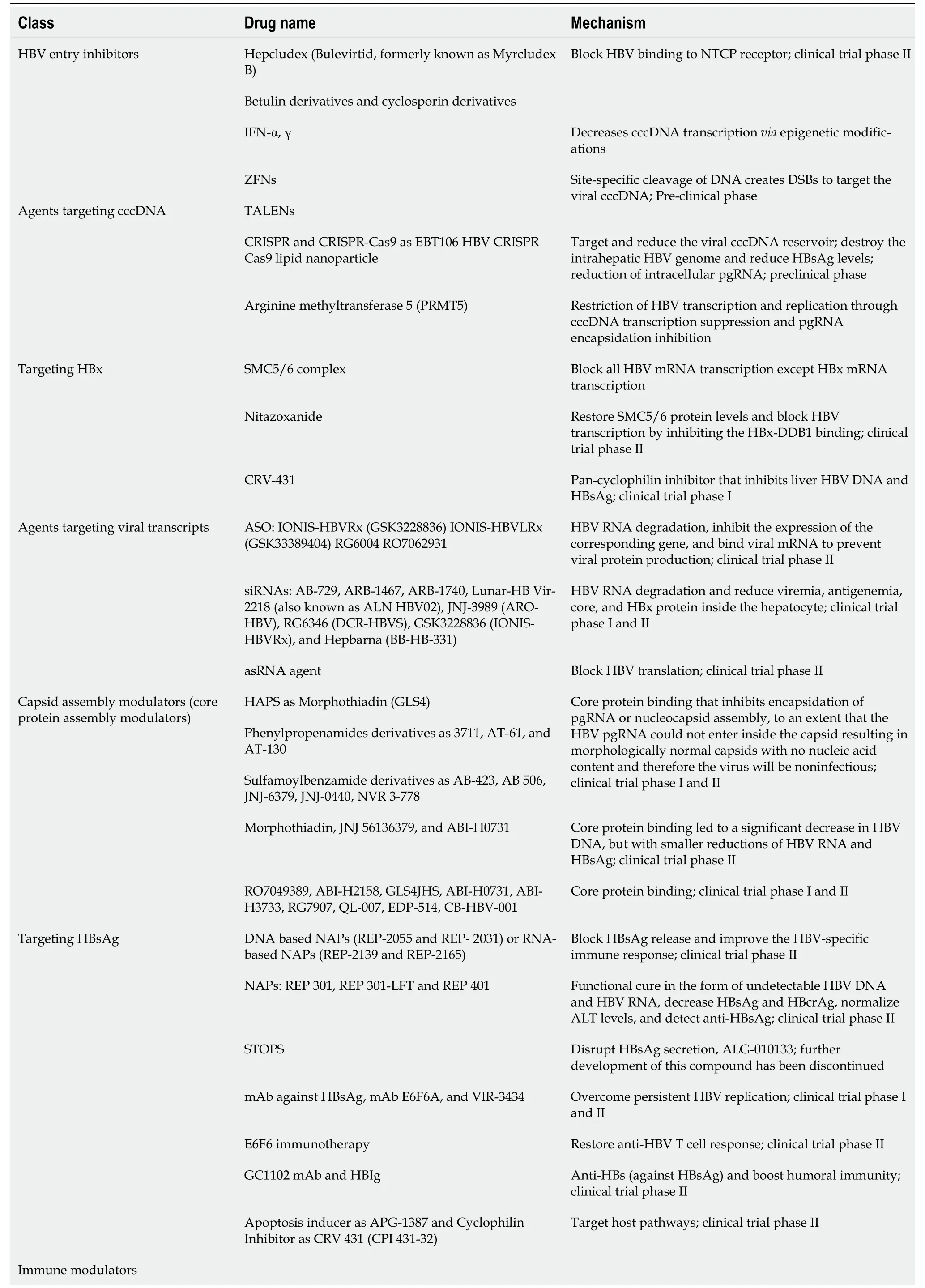
Table 2 New hepatitis B virus treatment modalities and clinical trial phases
Agents that directly target the virus
The direct-acting antiviral (DAA) therapy should be non-toxic; however, off-target effects may inevitably occur. Clinical trials have been carried out on novel NAs targeted to overcome the drawbacks of the currently approved NAs. Besifovir, a prodrug of tenofovir comprised of tenofovir exalidex and tenofovir disoproxilorotate, is currently under investigation[75-78]. The following are the novel DAAs that are currently in experimental or clinical trials.
HBV entry inhibitors
HBV enters the hepatocyte through binding irreversibly to the Na+-taurocholate co-transporting polypeptide (NTCP), which is a receptor exclusively expressed on hepatocytes with high-affinity to HBV and hepatitis D virus (HDV). The myristoylated preS1 domain (myr- preS12-48lipopeptide) of the LHB is important for this binding complex, triggering viral endocytosis[79]. It is believed that de novo infection through the NTCP receptor is required to maintain the cccDNA pool. Hepcludex (bulevirtid,formerly known as myrcludex B) is a synthetic analogue of the myr-preS12-48lipopeptide and specifically blocks NTCP HBV/HDV binding. It is in a clinical phase II trial[80].
They found that bulevirtide monotherapy or with PEG-IFN-α resulted in a significant, undetectable level of HDV RNA within 24 wk of treatment. For these results, it was approved for HDV treatment by the European Medicine Agency[81,82]. On the other hand, it was not expected that bulevirtide would provide a sustained decrease of hepatocyte HBV DNA levels, especially when used as monotherapy[83]. The reported adverse effect for this drug was interfering with the physiological function of NTCP(reuptake of circulating bile acids from the portal blood into the liver), leading to the elevation of plasma bile acids[84]. Further studies are required before bulevirtide can be applied for the treatment of HBV infection[85].
Betulin derivatives and cyclosporin derivatives were found to have a promising potent and selective inhibition of NTCP to HBV without affecting its role as a bile acid transporter[86]. Neutralizing antibodies are another promising entry inhibitor directed toward the antigenic S, preS1-domain, cellular heparan sulphate proteoglycans (e.g.,poly-Lysin), or attachment inhibitors that bind the virus (e.g.,heparin)[80].
Rather than targeting the receptor, several monoclonal/polyclonal antibody preparations are being developed that bind to the N-terminal region of pre-S1, the site of viral interaction with NTCP[87]. In addition to blocking viral entry, monoclonal antibodies can lower viremia and the level of subviral particles and may cross-present viral antigens with stimulation of T cells leading to HBsAg loss[67].
Agents targeting covalently closed circular DNA
In the hepatocyte nucleus, the relaxed circular DNA genome is converted into cccDNA, the minichromosome for HBV transcription and replication, resulting in persistence of HBV infection. IFN-α decreases cccDNA transcriptionviaepigenetic modifications[88]. Gene therapy including editing,silencing, or epigenetics are currently assessed to modify HBV, most of them aiming to permanently disable cccDNA and lead to HBV infection cure.
Gene editing introduces genome targeted modifications using engineered nucleases for cutting a specific genomic target sequence[89]. They induce double strand breaks (DSBs) at a particular site in the HBV genome resulting in activation of the repair mechanisms of cellular DNA, aiding in producing targeted genome modifications at specific areas[90]. Three classes of nucleases have been developed:zinc finger nucleases (ZFN); transcription activator-like effector nucleases (TALENs); and the clustered regularly interspaced short palindromic repeats (CRISPR) system[91]. They have the ability to make DSBs and target cccDNA. The DSBs are repaired by nonhomologous end-joining pathways, producing insertions and deletions and disrupting the genes open reading frames. Moreover, they lead to degradation of cccDNA[92-94].
ZFNs are designed with zinc finger domains and Fok1 nuclease domain. The zinc finger domains bind to specific sites on DNA, which facilitate the introduction of a DSB into a specific targeted locus. It stimulates gene targeting 100-fold to 10000-foldviathe activation of DNA repair mechanisms[95,96].However, ZFN production needs extensive protein engineering, and for every new genome target a new ZFN must be created. Moreover, its production is time-consuming and costly. Therefore, it is not an attractive therapeutic agent[97].
TALENs are superior to ZFNs, regarding simplicity. Instead of binding to three nucleotides, they only bind to one nucleotide, executing an increased specificity with a decline in the off-target effects.TALENs use a catalytic or functional domain of a nonspecific FokI endonuclease, which can effectively induce the formation of DSBs to the target[98,99]. The DNA binding domains of TALENs are the repeat region of transcriptional activator-like effector protein[100].
CRISPR and CRISPR-associated protein nine nuclease (Cas9) is produced fromStreptococcus pyogenes[101]. It directly targets and reduces the viral cccDNA reservoir. CRISPR/Cas9 is simply formed of Cas,and it precisely allows gene sequence editing with specific cleavage of cccDNA[75]. Unfortunately, it bears the risk of host genomic rearrangement and damage when targeting the integrated HBV DNA causing DSBs of the host genome[102,103]. The CRISPR/Cas9 approach revealed a potent effect in destroying the HBV genome and reducing HBsAg levels bothin vitroand in animal models[104]. It is more efficient and more cost-effective than ZFNs and TALENs[62]. However, this strategy has several unwanted effects such as accurate targeting of the DNA site, off-target results on the host genome,mutation introduction, and resistance development towards the Cas9/single guide RNA system[105,106].
Targeting HBV DNA using HBV-infected HepG2-NTCP cells with CRISPR-Cas9 resulted in cleavage and the subsequent appearance of episomal HBV DNA variants. The effects of Cas9 were sustainable,signifying permanent changes in the HBV genome (not just transient effects owing to transcriptional interference)[107].
A novel technology of the CRISPR system has been developed to allow permanent damage to the HBV genome and finally reach a complete cure. This was accomplished by producing premature stop codons, without causing a DSB of the host genome[108]. Persistent high levels of HBsAg are considered a critical risk factor for the development of HCC.In vitroexperiments of the CRISPR/Cas9 system showed significant functional inactivation of cccDNA by mutagenesis, inducing suppression of viral replication and potent synergy with NAs[109,110]. Results suggest that additive modulation of host DNA repair pathways enhances the antiviral activity of CRISPR/Cas9 and causes profound reduction of intracellular pgRNA[111]. However, fragments of viral DNA appear to be repaired in transcriptionally active “small cccDNA” formations[110]. The HBV DNA variants were generated after dual pgRNA targeting, highlighting the importance of understanding the fate of cccDNA after gene editing.
It is expected that cccDNA transcription is controlled by epigenetic machinery, mainly through cccDNA-bound histone 3 and histone 4 acetylation. DNA epigenetic modification could silence gene expression. Epigenetic modifications include cccDNA methylation and acetylation. HBV transcription and replication can be effectively restricted by protein arginine methyltransferase 5. HBV replication is limited by protein arginine methyltransferase 5 through cccDNA transcription suppression and pgRNA encapsidation inhibition[111-113]. Epigenetic editor studies are still preliminary for assessing its effect on HBV replication and sustainability[53].
A recent study revealed that non-histone host DNA-binding protein high mobility group box 1 targets the HBV cccDNA minichromosome and induces epigenetic silencing of viral transcription,which could be antagonized by the viral HBx protein[114]. Base editors are alternative gene editing tools that were engineered by fusing partially inactivated Cas nucleases to deaminases. They induce accurate mutations in the targeted sequencesviadeamination reactions, without promoting DSBs. This technique does not require donor DNA templates for gene correction[107]. The key to curing CHB infection is to eliminate or silence cccDNA within the hepatocyte nucleus. Therefore, there are several approaches in preclinical development.
Agents targeting HBx
The structural maintenance of chromosomes protein 5 and 6 (SMC5/6) complex binds to the episomal cccDNA templates. Through this binding, the SMC5/6 can block all HBV mRNA transcription except HBx mRNA transcription. In the cytoplasm, the HBx protein binds to the cullin 4 RING E3 ubiquitin ligase complexviaits interaction with the cellular damaged DNA binding protein 1 (DDB1) forming the DDB1-CUL4 complex[115,116]. This complex is then transported to the nucleus targets to destroy the cccDNA-bound Smc5/6 complex, allowing cccDNA transcription[117]. In addition, it promotes development of HCC[118]. Accordingly, antiviral agents that interfere with HBx action can also lead to silencing of cccDNA transcription[119]. In cell culture models, nitazoxanide was found to restore SMC5/6 protein levels and block HBV transcription by inhibiting the HBx-DDB1 binding[120]. In a pilot study, nitazoxanide was given to nine treatment-naïve CHB patients for 48 wk. Results revealed undetectable HBV DNA in 89% of patients, and HBsAg disappeared in 33% of patients. HBeAg seroconversion occurred in the 2 HBeAg-positive patients. The drug was well tolerated with mild to moderate transient side effects that resolved during treatment. However, there was no follow-up for the responders[121].
Pevonedistat, a neuronal precursor cell expressed developmentally, downregulated protein 8 activating enzyme inhibitor and dicoumarol, an inhibitor of NAD(P)H quinone oxidoreductase 1, which was shown to reduce HBx expression[122,123], restore SMC5/6 levels, and suppress viral transcription in cultured hepatocytes[124] in a humanized mouse model. However, the major limitation to this approach may be the reactivation of cccDNA as soon as HBx becomes available again[122].
Agents targeting viral transcripts
Viral RNA is the basis of the viral antigens and proteins. Prevention of viral replication and HBsAg production can be achieved through RNA inhibition of viral transcripts translation. This might restore the HBV-specific immune response and potentially lead to a functional cure. RNA interference (RNAi)through small-interfering RNA (siRNA) and anti-sense oligonucleotide (ASO) are new strategies for CHB treatment. RNAi targets post-transcriptional mRNAs and pgRNAs to decrease both HBV antigen production and viral replication. Reducing viral antigens may promote host immune reconstruction against HBV[125]. siRNA and ASO have different mechanisms of gene silencing. The double-stranded siRNA enters the cell cytoplasm, the duplex opens, and the antisense strand binds to the RNA-induced silencing complex (RISC). The antisense-RISC complex recognizes the mRNA. After binding, RISC induces gene silencing and mRNA degradation[126]. RISC is defined as a multiple turnover enzyme; a single siRNA could silence multiple mRNA transcripts after being induced by RISC[127]. However,single-stranded DNA oligomers (ASO) enter the cell and the nucleus, and they can bind to target mRNA alone. It binds to its corresponding site on the target viral RNA forming a DNA-RNA duplex. This allows HBV RNA degradation by ribonuclease H (a family of endonucleases), resulting in inhibition of the corresponding gene expression[128].
Small interfering RNAs (siRNAs) are designed to lower serum HBsAg through targeting specific viral mRNA sequences for all HBV RNA levels, even HBx mRNA[129]. Thus, siRNAs permit lowering viremia, antigenemia, and reducing the core and HBx proteins inside the hepatocyte. Several agents have been developed and reached phase II clinical trials, including the siRNAs VIR-2218 (also known as ALN HBV02), JNJ-3989 (ARO-HBV), RG6346 (DCR-HBVS), and JNJ-3989 (ARO-HBV) and the ASO GSK3228836 (IONIS-HBVRx)[130]. Studies are currently assessing if restoration of SMC5/6 complex induced by siRNA can lead to cccDNA silencing[119,131]. In a phase II study, a single dose siRNA ARC-520 in combination with entecavir resulted in a profound and durable decrease in serum HBV DNA and HBsAg levels[42]. In phase I/II clinical trials, re-designing siRNA to be able to target both integrated and cccDNA viral sequences resulted in an effective decrease in serum HBsAg levels in both HBeAgpositive and HBeAg-negative patients[132].
Several siRNA drugs are in phase II clinical trials. In 98% of the participants, NA co-administration with JNJ-3989 induced a decrease in HBsAg more than 1.0 Log10 IU/mL and a decrease of 1.93 Log10 IU/mL[133]. AB-729, Vir-2218, and RG6346 showed similar reductions in HBsAg titer[134]. An ASO phase II clinical study was conducted for GSK3228836. Each group of treatment-naïve and previously NA-treated received 300 mg six times. The serum HBsAg titer mean was reduced by more than 1.5 Log10 IU/mL[135]. The risk of post-treatment reactivation by the remaining cccDNA, the potential toxicity of the delivery vehicle, and the off-target toxicity are the main topics of interest for these agents.
Capsid assembly modulator inhibitors
The HBV core protein or HBcAg is essential for genome packaging, reverse transcription, and potentially for modulation of cccDNA. The HBc has recently emerged as a promising direct antiviral target that would affect multiple aspects of the viral life cycle. Several small molecules (chemical classes)are named core protein assembly modulators (CpAMs). There are two main classes of CpAMs. Class I are known as heteroaryldihydropyrimidines. They target the core protein and alter the kinetics of nucleocapsid formation resulting in deformed virus particles that would be noninfectious. Class II includes phenylpropenamides and sulfamoylbenzamide derivatives. They speed up capsid assembly to an extent that the HBV pgRNA could not enter inside the capsid resulting in morphologically normal capsids with no nucleic acid content and are therefore noninfectious[136,137]. Several CpAMs are in preclinical and clinical studies including morphothiadin, JNJ 56136379, and ABI-H0731[50]. They led to a significant decrease in HBV DNA but with smaller reductions of HBV RNA and HBsAg[138,139].Thus, CpAM compounds directly revoke viral replication and post-infection spread[140].
In a preclinical study, adding GS-SBA-1 (a CpAM-E class, which is a potent inhibitor of extracellular HBV DNAin vitro) at the time of infection inhibited cccDNA establishment and the downstream viral products (HBsAg, HBeAg, and viral RNA).In vitro, GS-SBA-1 avoids creation of extracellular HBV RNA-containing viral particles. It is a potent CpAM that improved viral suppression when combined with an NA[141]. In clinical trials, the combination of NAs and CpAMs might lead to more potent suppression of viral replication, which could decrease the renewal of the cccDNA and inhibit its formation in newly infected cells[142]. However, withdrawal of the CpAM and NAs after short-term administration resulted in minimal changes in HBeAg and HBsAg levels coupled with a high rate of virologic relapse[143].
HBsAg release inhibitor
HBsAg enhances viral entry through binding of the preS1 region to the NTCP receptor. HBsAg is the most abundant HBV antigen, representing about 99.99% of circulating SVPs. This high circulating HBsAg contributes to the suppressive immune environment through interference with the signaling pathways of innate and adaptive immunity, contributing to chronicity of HBV[144]. Inhibition of HBsAg release is a potential step in preventing the release of enveloped HBV and the spread of infection and allowing improvement of the HBV-specific immune response. In addition, it clears circulating HBsAg in patients causing persistence control of HBV infection even after therapy withdrawal[124].
Nucleic acid polymers (NAPs) reduce the release of HBsAg through interrupting the assembly and secretion of HBV subviral particles. They are single-stranded nucleotides. They are either DNA-based NAPs (REP-2031 and REP- 2055) or RNA-based NAPs (REP-2165 and REP-2139). In the REP 102 study by Al-Mahtabet al[145], REP-2139-Ca monotherapy was given to 12 Bangladeshi patients; 9/12 patients showed 2.79–7.10 Log reduction of HBsAg. After a combination with PEG-INF-α or thymosin alpha 1 immunotherapy, there was a disappearance of HBsAg. Adverse effects occurred primarily during combination therapy. They were in the form of loss of appetite, dyspepsia, fever, and hair loss.
A phase II clinical trial of REP 2165 and REP 2139 revealed that the combination of either NAP with tenofovir TDF and PEG-IFN-α significantly decreased HBsAg levels in 15/20 patients, with seroconversion occurring in 11/20 patients with CHB in the first 24 wk. However, hepatitis flares were more common among patients receiving NAPs, especially those with undetectable HBsAg, suggesting that host-induced flares are the result of immune control of infection[146]. In clinical trials, a functional cure was developed after receiving REP 301, REP 301-LFT, and REP 401[147]. These effects of NAPs need to be confirmed in larger studies with monitoring the long-term safety and efficacy.
A monoclonal antibody (mAb) against HBsAg, mAb E6F6A, revealed a significant effect in a mouse model to overcome persistent HBV replication[148]. Another study reported restoration of anti-HBV T cell response with E6F6 immunotherapy in mice[149]. Recently, GC1102 mAb against HBsAg was investigated in a phase II clinical trial[80].
Like NAPs, S-antigen transport-inhibiting oligonucleotide polymers (STOPS) are single-stranded oligonucleotides that sequester cellular proteins necessary for HBsAg production and disrupting HBsAg secretion. STOPS were shown to be more potent than NAPsin vitro. STOPS could contribute to the functional cure of CHB as it inhibits the production of HBsAg and HBeAg and accelerates degradation of HBsAg. Antiviral compounds that act on cellular proteins will decrease the chance of the appearance of viral resistant mutations[71]. However, these molecules acting on cellular targets may exhibit toxicity.A phase I study evaluating the STOPS agent ALG-010133 demonstrated no reduction of HBsAg.Therefore, its further development has been discontinued[150].
Immune modulators
To overcome acute HBV infection, the adaptive immune responses are the cornerstone for viral resolution through B and T viral specific cell function activation. Failure of the host immune response results in liver damage and CHB[151]. In CHB, the CD8+ T cells show functional impairment in both antigen-specific cytolytic activity and non-cytolytic functions, as the secretion of IFNγ and tumor necrosis factor-alpha are needed for intracellular viral elimination through killing the infected host cell[152]. High HBsAg load also leads to T cell functional exhaustion and apoptosis[153]. NK cells in the blood and liver have additional killing and depletion effects on HBV-specific CD8+ T cells[154]. In CHB,surviving HBV-specific CD8+ T cells are markedly dysfunctional compared to that in acute HBVresolved patients[155].
Endosomal toll-like receptors (TLR) and cytosolic retinoic acid inducible gene I (RIG-I), melanoma differentiation antigen 5, or cyclic GMP-AMP synthase recognizes intracellular pathogenic nucleic acids leading to cytokine production and transcription of IFN-stimulated genes. HBV is able to evade and suppress the recognition of innate immunity[156]. An innovative immunotherapy approach is targeting functional restoration of T cell[151]. TLR agonists, RIG-1 agonists, checkpoint inhibitors, monoclonal antibodies, genetically engineered T cells, and therapeutic vaccines are being discovered.
Therapeutic vaccination
The concept behind the therapeutic vaccines is the introduction of modified HBV antigens that will interact with antigen-presenting cells. The antigen-presenting cells stimulate HBV-specific T cells to produce antiviral cytokines such as IFN-γ[157]. Preclinical data in HBV mice models revealed good efficacy with therapeutic vaccines. However, in humans the results were unsatisfactory. This may be attributed to the high level of HBsAg in CHB patients, which interferes with the ability of the vaccine to induce HBV-specific T cells. GS-4774, a yeast-based T cell vaccine, was the first HBV therapeutic vaccine studied in HBV-infected humans. It was safe, well-tolerated, and increased cytokine production.However, there was no significant decline in HBsAg by week 48[158,159].
The therapeutic vaccine BRII-179 was evaluated in a phase I/IIb trial. It is a virus-like particle encoding for the three HBsAg proteins. BRII-179 produced a good humoral response in all vaccinated patients, and it increased the frequency of IFN-γ-producing T cells. However, there was no significant decrease in the level of HBsAg, HBV RNA, or HBcrAg[160]. To enhance the effectiveness of therapeutic vaccines, a combination with antiviral approaches that induce a satisfactory immune response are ongoing. This includes therapeutic vaccines with checkpoint inhibitors, TLR or RIG-I agonists, and/or other novel therapy to allow viral antigen clearance[80].
Checkpoint inhibitors
T cells of patients with CHB infection overexpress inhibitory receptors such as cytotoxic T lymphocyte associated antigen 4, programmed cell death protein 1 (PD-1), and T cell immunoglobulin and mucin containing 3. This may affect the T cell function resulting in decreasing the T cell immune response[153,161-163]. Blocking these checkpoint inhibitors may recover the exhausted T cells and restore their activity. T cell immunoglobulin and mucin containing 3 and cytotoxic T lymphocyte associated protein 4 checkpoint blockade were able to restore virus-specific CD8+ T cell responses in CHB patients[164,165].Anin vitrostudy was carried out on HBV-specific CD4+ T cells from CHB with HBeAg-negative infected persons. There was a functional boost of HBV-specific CD4+ T cells combined with programmed death ligand 1 blockade, leading to a marked increase in interleukin (IL)-21 and IFN-γ production. After blocking IL-10 receptors there was a reverse immune dysfunction in CHB infection[166]. Anotherin vitrostudy using anti-PD-1/anti-programmed death ligand 1 antibodies revealed only mild restoration of both peripheral and intrahepatic HBV-specific T cells[167]. However, in a preliminary clinical trial only 1 out of 12 patients achieved HBV cure after receiving minimal doses of anti-PD-1 antibodies[168].
Among the non-responder CHB patients, IL-2 therapy was able to significantly increase the frequency and function of HBV-specific CD8+ T cells, which was associated with an improvement in clinical outcomes and HBeAg seroconversion. It was concluded that the sequential IL-2 therapy has an effect on liberating the immune function in refractory CHB patients[169]. Another study revealed that HBVspecific T cells from immune-tolerant HBV-infected patients were liberated by IL-2 treatment, with high levels of IFN-γ, which was similar to immune-active patients. They concluded that a modified IL-2 molecule has the ability to reduce the toxic effects on T cells and are only directed towards CD8+ T cells[170].
However, the use of checkpoint inhibitors in the clinic has been constrained due to the death of hepatocytes causing acute liver failure and risk of autoimmunity[67]. The use of checkpoint blockade in CHB infection may be limited, despite their potential usefulness due to their safety and unpredictable response. Therefore, future safety studies are expected.
Toll-like receptor agonists
Several receptors, defined as pathogen recognition receptors, are required for effective function of the innate immune cells. The pathogen recognition receptors include TLRs, RIG-I-like receptors, formylmethionyl-leucyl-phenylalanine, and others[171]. These receptors are the primary sensors of infection,and they represent the essential component for innate immune responses[12]. TLRs are the first line of defense against invading pathogens. They are accountable for detecting self and non-self-antigens,stimulating the maturation of dendritic cells, and initiating antigen-specific adaptive immune responses[172,173].
TLR agonists (TLR-7 and TLR-8) initiate the manufacture of endogenous IFNs, the induction of IFNstimulated genes, and the activation of other signaling cascades such as the Janus kinase/signal transducer and activator of transcription signaling pathway, leading to HBV replication inhibition[174].GS-9620, a TLR-7 agonist, increases the T cell and NK cell responses, while it reduces the capability of NK to suppress T cells[175]. GS-9620 achieved a durable suppression of HBV replication in preclinical studies. It allowed the induction of type I IFN[176]. In HBV-infected chimpanzees, oral GS-9620 induced a decrease in circulating levels of HBsAg and HBV DNA in the serum and hepatic cells[177] as well as in woodchucks[176]. In a phase II study, GS-9620 was given once weekly and was safe and well-tolerated in CHB patients who were virally suppressed by oral antiviral treatment. The study revealed a regular dose-dependent pharmacodynamic induction of IFN-stimulated gene mRNA expression. However,HBsAg levels did not significantly decline[178]. It does not produce antiviral effects despite the stimulation of host NK and HBV-specific T cell responses[175].
Adding to its ability for immunomodulation, TLR stimulation was found to directly stimulate T cell function through metabolic regulation[179]. TLRs play an important role in sensing the initial invasion and activation of the innate immune response. In a phase Ia and phase II trial, selgantolimod (GS-9688),an oral TLR-8 agonist, enhanced the production of antiviral cytokines in preclinical studies and was used with no significant decline of HBsAg in virally suppressed patients with CHB and viremic CHB[180,181].
Genetically engineered T cells
In order to strengthen HBV-specific T cell responses (overcome HBV-specific T cell exhaustion), anin vitrogenetically engineered T cell carrying a chimeric T cell receptor containing anti-HBs-specific antibody or an HBV-specific T cell receptor gene transferred through a vector have been developed. In animal models andin vitrostudies these engineered T cells were able to recognize HBV-infected cells,carrying HBV proteins on their surfaces resulting in disappearance of HBV-infected cells and decreasing cccDNA[182,183]. They induced HBV infection clearance through cytokine secretion and cytotoxicity[184,185]. It has the advantage that the T-cell receptor (TCR)-T cells are not suppressed by the high levels of soluble antigens in the serum of CHB patients, as they are not recognized by them[186].
However, there are some concerns about the safety of this technology, as it carries the risk of going beyond immune control resulting in liver damage, and this limited its clinical use. Therefore, the addition of potent antiviral treatments to selective immune modulation may be a good strategy inducing functional cure for HBV, without causing severe liver damage progression[187]. TCRreprogrammed non-lytic T cells have been developed to avoid the risk of severe liver damage. It can produce low amounts of perforin and granzyme B. However, in HBV-infected hepatocytes it is able to decrease viral infection by activation of the anti-viral cytidine deaminases APOBEC3[187,188]. Despite several studies currently addressing engineered T cell therapy challenges, its clinical application is still limited as it is technically difficult.
iC9 and HSV-TK were assessed as safety systems in the context of adoptive T cell transfer for the treatment of persistent CHB[189].In vitroT cell cytotoxicity and cytokine production were abruptly stopped after HBV-specific TCR and S-chimeric antigen receptor T cells were infected with iC9 or HSVTK.In vivoS-chimeric antigen receptor T cell induction of iC9 resulted in a rapid and significant decrease of transplanted T cells and prevented cytokine release and liver damage. However, it resulted in a decrease of antiviral effectiveness.
Preliminary evidence in an animal model demonstrated the reduction of HBsAg and HBV DNA levels without inducing significant liver damage. To prove the safety and efficacy of using genetically engineered T cells, an HBsAg-specific TCR or adoptive transfer of autologous T cells expressing HBVspecific TCR were demonstrated in patients with HCC caused by HBV[190,191]. To better understand the safety and efficacy of these novel approaches, results from clinical studies in non-HCC patients are necessary.
COMBINATION THERAPY
In theory, triple combinations of the three categories of therapy (reduction of viral antigen load,inhibition of viral replication, and immune stimulation) might be considered the ideal combination. To inhibit transcription and replication and boost immunity, vonafexor is a new agent. It is a farnesoid X receptor agonist that reduces HBV transcription. In a phase II open label study, HBeAg-negative patients with CHB were treated with TDF for 24 wk and randomly allocated to receive 24 wk of a NAP(REP 2139 or REP 2165) plus TDF and PEG-IFN or PEG-IFN plus TDF to reduce antigen burden, inhibit replication, and boost immunity[146].
The data available recommend combination therapy to achieve functional cure in CHB[192]. The best clinical evidence of the benefits of the alternative therapy is the combination of NAs and PEG-IFN,which were examined in a meta-analysis. Initial combination therapyvsinitial NA monotherapy showed a nonsignificant increased relative risk of HBsAg loss of 1.44. While PEG-IFN add-onvsNA monotherapy showed a significantly improved relative risk of 4.52[193].
A recent study evaluated the combination of siRNA (JNJ-3989) with or without a CpAM (JNJ-6379)plus NA. It revealed that the triple regimen (siRNA plus CpAM plus NA) had the lowest rate of response compared with siRNA plus NA only[194]. This raises the possibility of an interaction between the CpAM and siRNA. Therefore, not all combinations will result in synergy.
Kuiperyet al[195] treated infected HepG2-NTCP with TDF, TLR 7/8 agonists, or RNAi using siRNAs.They found that TDF and TLR7/8 agonists had no impact on T cell recognition. The administration of siHBVs, which inhibit viral replication and antigen expression, strongly suppressed HBV-specific CD8 T cell recognition. Similarly, we demonstrated that siHBV-mediated antigen reduction could negate the ability of TLR conditioned media to enhance antigen presentation to HBV-specific CD8 T cells.Understanding how CD8 T cell recognition is altered by these drug classes will help inform logical combination strategies for hepatitis B treatments. Immunomodulation and RNAi, but not NAs, alter the recognition of infected HepG2-NTCP by HBV-specific CD8 T cells. This could present a significant obstacle for combining immunotherapies and require careful immunological follow-up post-RNAi treatment.
ASSESSMENT OF SAFETY AND INDICATIONS FOR STOPPING TRIALS OF NEW HBV THERAPIES
The safety of new HBV therapies should always be compared with the well-proven safety of NAs.Increased additional risk should be weighedvsthe predictable clinical outcomes. ALT flares are a major concern in HBV drug application, which may be induced either in relation to host immune responses or antiviral or drug-induced flares. Those related to the host immune response are associated with a decrease in serum HBV DNA and viral antigen levels. Virus-induced flares may be related to increased viral activity, lack of drug efficacy or resistance, or the side effects of antiviral agents[196]. Development of ALT flares necessitate close patient monitoring to identify the type of flare, whether to stop the antiviral, and whether it can be resumed. Even after ceasing treatment, close monitoring is mandatory,as flares may also occur due to viral reactivation, immune recovery, or delayed liver toxicity.
WORLDWIDE ELIMINATION OF HBV
Hepatitis B elimination is achievable but needs greater financial support. It will not be attained without finding and treating all HBV-infected people. In developing countries, many people are unaware of their infection, and few are receiving antiviral therapy[197,198]. The goals are to reduce new infections by 90% by 2030, with a prevalence in children of no more than 0.1%, by improving the delivery of a birth dose of vaccine, which will increase from 39% in 2015 to 90% globally. In addition, all pregnant women will be tested for HBV, and new antiviral treatment-based interventions will be developed[199].The vaccine is highly cost–effective, safe, and available as a combination with other vaccines in the Expanded Program on Immunization. Universal hepatitis B vaccination should be undertaken in all countries regardless of HBV endemicity. The hepatitis B vaccine is the first “anti-cancer” vaccine, as it prevents hepatitis B, the leading cause of HCC[200]. The introduction of HBV immunization in 189 countries by the end of 2018 resulted in an estimated 84% of the world’s population receiving three doses of the HBV vaccine. Nonetheless, the estimates are still insufficient (76%) in Africa, and just 11% of newborn babies in Africa received the HBV birth dosage, which is still a very low coverage rate (38%)internationally[199].
In Egypt, birth dose has been nationally added to the HBV vaccination program at the beginning of 2017[6]. After vaccination, anti-HBs levels decline over time[201]. Immunocompetent people who achieve an anti-HBs level ≥ 10 mIU/mL after receiving three doses of the hepatitis B vaccine remain protected, even if anti-HBs levels decline to < 10 mIU/mL due to persistent memory cells. Booster doses are not recommended for people with normal immune status who have been vaccinated[202-204].Revaccination may be recommended for non-responders and certain high-risk populations[5,205].
Intense viral deactivation alone does not attain HBsAg reduction or loss. Combining NAs and immunotherapy reduces HBsAg levels and induces HBsAg loss in some patients, particularly those with low baseline HBsAg levels. Agents that are specifically prepared to decrease viral antigen load could not achieve constant HBsAg loss when used alone. Thus, the use of combinations of all three therapy types is recommended[142]. Moreover, combination therapy with the current anti-HBV therapy would be more effective in causing a drop in HBsAg levels and the eventual curing of HBV. To evaluate the synergistic effect of anti-HBV drugs and eradicate this global health issue, combination therapy with CpAM and entry inhibitors, siRNA, immunomodulators, or therapeutic vaccines to rebuild the immune system may be used[140].
CONCLUSION
HBV persistence can occur in an overt state with the presence of serum HBsAg due to induction and maintenance of a defective immune response, especially an adaptive immune response resulting from the interaction between viral and host factors. It can also persist in a long-term occult state with the absence of serum HBsAg, owing to the long life of the HBV genome, including cccDNA and integrated DNA within the nucleus of hepatocytes. Nine medications are now licensed for the treatment of CHB,including two IFN conventional and PEG-IFN formulations as well as seven NAs (lamivudine,telbivudine, adefovir, entecavir, TDF, tenofovir alafenamide +, and besifovir dipivoxil) (only in Korea).The therapeutic vaccines that have been studied to date are largely ineffective at significantly and permanently suppressing HBV in animal models or CHB patients, but they are quite successful at priming particular T and B cell responses to HBV antigens.
Although a functioning T cell response is necessary for the management of HBV, it is unquestionably insufficient for an effective immunotherapeutic strategy. Gene therapy including editing, silencing, and epigenetic alterations have been assessed to modify HBV. Most of them act through direct deactivation of cccDNA and HBV infection cure. Monotherapy of entry inhibitors is not expected to induce complete elimination of cccDNA due to a slow rate of hepatocyte turnover and cccDNA decay. In order to effectively treat CHB, a combination of NA-based antiviral therapy, intrahepatic innate immunity activation, stimulation of particular T cell responses, and activation of non-immune mechanisms for sustained HBV control without unfavorable side effects may be required. The combined use of potent antiviral treatments and selective immune modulation may be the best strategy to accomplish a functional cure for HBV, without inducing severe tissue damage and disease progression. To reach the goal of global elimination of HBV by 2030, the WHO published certain measures that include full childhood vaccination, preventing perinatal transmission, harm reduction, blood safety, and testing and treatment. Unfortunately, most countries are not on track to meet these targets by 2030.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Eng. Salma El-Atroush for designing and drawing the figure presented in this review article and Omer A Kamal.
FOOTNOTES
Author contributions:Salama II designed the review and implementation; Salama II, Salama SI, Abdel-Latif GA, and Raslan HA were responsible for writing the manuscript; Sami SM, Shaaban FA, Abdelmohsen AM, and Fouad WA were responsible for the first review; and all authors reviewed and approved the manuscript.
Conflict-of-interest statement:All authors report having no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Egypt
ORCID number:Iman Ibrahim Salama 0000-0001-8901-4625; Samia M Sami 0000-0003-1832-2932; Somaia I Salama 0000-0001-8013-2342; Ghada A Abdel-Latif 0000-0001-7165-0594; Fatma A Shaaban 0000-0003-4129-2727; Walaa A Fouad 0000-0001-9687-8245; Aida M Abdelmohsen 0000-0003-4918-8967; Hala M Raslan 0000-0002-7571-5241.
S-Editor:Liu XF
L-Editor:Filipodia
P-Editor:Cai YX
杂志排行

World Journal of Hepatology的其它文章
- Cerebrospinal fluid liver pseudocyst: A bizarre long-term complication of ventriculoperitoneal shunt: A case report
- Giant cavernous hemangioma of the liver with satellite nodules:Aspects on tumour/tissue interface: A case report
- Liver steatosis in patients with rheumatoid arthritis treated with methotrexate is associated with body mass index
- Respiratory muscle training with electronic devices in the postoperative period of hepatectomy: A randomized study
- Current guidelines for diagnosis and management of hepatic involvement in hereditary hemorrhagic teleangiectasia
- Fatty liver and celiac disease: Why worry?