沿Blaschko线分布的线状汗孔角化症1例
2023-05-12王莎吕静江阳李佳佳
王莎,吕静,江阳,李佳佳
1 临床资料
患者男,16岁,左侧躯体线状角化性丘疹、斑块13年。患者自幼发病,3岁时先于左侧面颊部出现孤立的圆形角化性丘疹,皮疹缓慢增大增多,青春期进展明显,沿左侧躯体Blaschko线出现了线状角化性丘疹、斑块,周围堤状隆起,伴轻微瘙痒。除面部、耳廓、颈部出现皮损以外,患者左侧躯干、左侧上下肢、掌跖以及阴囊均有受累。为进一步诊治至本院皮肤科就诊。患者既往有前额左侧脂肪瘤病史、鼻部畸形史(已行手术治疗,具体不详)。皮肤科情况:左侧躯体见沿Blaschko线分布的淡红色角化性丘疹、斑块,中央轻度萎缩,边缘角化过度呈堤状隆起,表面少许鳞屑(图1)。左上肢皮损组织病理示:角化过度伴角化不全,可见柱状角化不全,其下方颗粒层减少或消失,可见角化不良细胞,真皮浅层炎细胞浸润(图2)。诊断:根据患者皮疹表现,结合皮损组织病理,诊断为线状汗孔角化症。治疗与预后:给予0.1%维A酸乳膏、糠酸莫米松乳膏外用皮损处,疗效欠佳,建议给予阿维A胶囊口服(患者及家属拒绝)。嘱定期随访以防止皮肤肿瘤的发生。
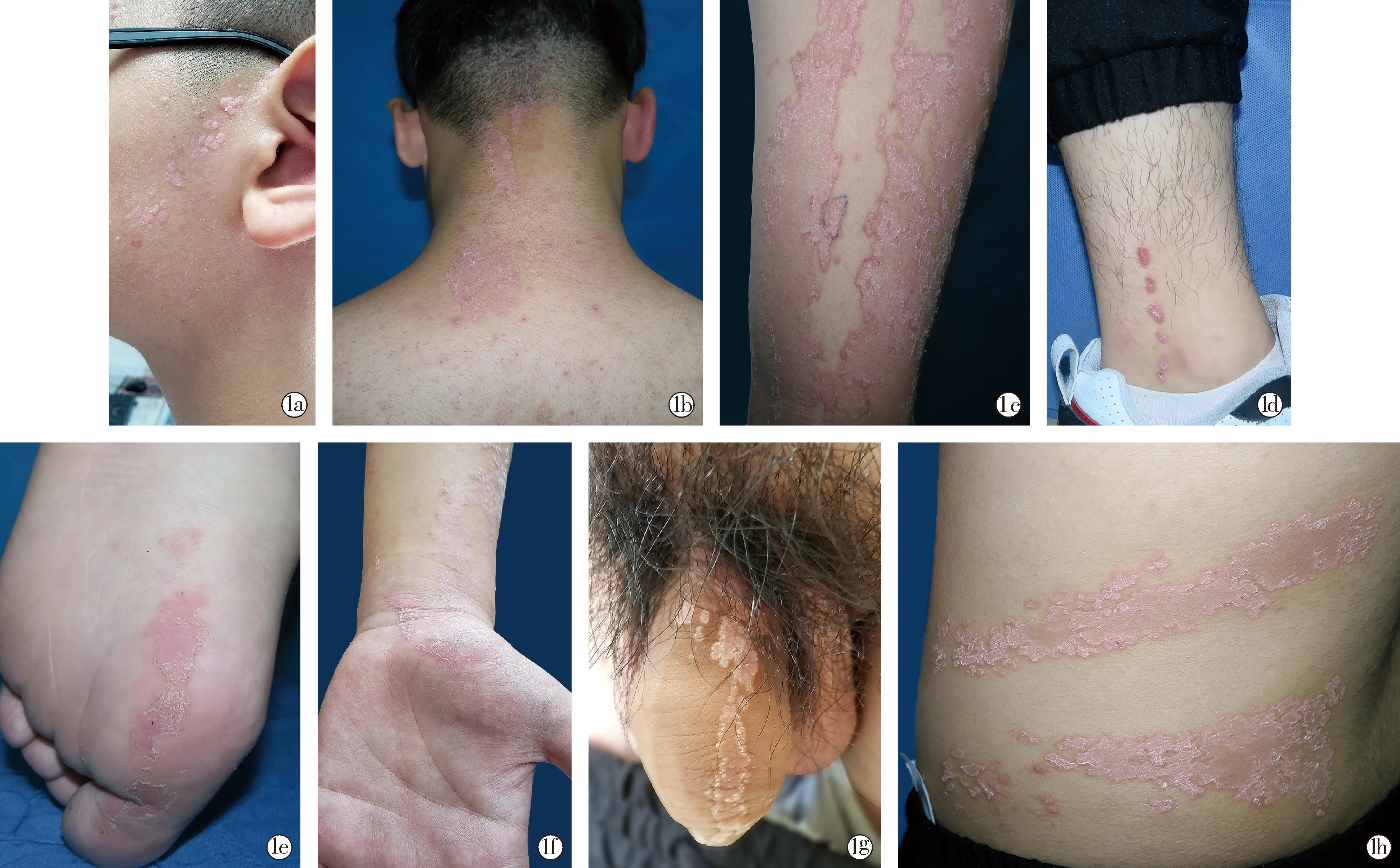
图1a~1h 临床图片:呈线状分布于左侧躯体的疣状丘疹和斑块,包括面部、颈部、躯干、上下肢、掌跖和生殖器,皮损边界清晰,表面疣状增生,中央轻度萎缩,边缘角化过度呈堤状隆起

图2a 角化过度伴角化不全,可见柱状角化不全,其下方见角化不良细胞,真皮浅层淋巴细胞浸润 (HE×40);图2b 角化不全柱和其下方的角化不良细胞 (HE×200)
2 讨论
汗孔角化症(porokeratosis,PK)是一种罕见的慢性角化不全性皮肤病,虽然该病主要以常染色体显性遗传方式遗传,但大多数病例为散发性[1]。单侧线状汗孔角化症是其中一种少见的亚型,沿Blaschko线分布生长发展的模式更为少见。
线状汗孔角化症是汗孔角化症的一种特殊类型,约占3.55%~16.7%,进一步分类包括局限性、带状、系统性或全身性[2],沿Blaschko线分布的鲜有报道。病变可能累及身体的任何部位,但生殖器[3]和面部[4]的病变都被认为是罕见的。该病为表皮细胞在某些刺激下(如免疫抑制、日光照射、慢性皮肤病等)的不正常克隆性增生,具有癌前倾向,免疫组织化学方法已经证明了皮损中P53蛋白呈过度表达[5]。本病基本皮损为角化性环形或不规则形棕褐色丘疹、斑块,中央略萎缩凹陷,周围呈堤状隆起[6],特征性组织病理可见角化不全柱,其下方部分颗粒层减少或消失,可见角化不良细胞[7]。线状汗孔角化症被列为一种罕见的疾病,它只发生在身体的某一部分,或全身性,当躯干受累时,皮损常沿Blaschko线呈带状分布[8-9]。本文报告的这例患者为青春期男孩,皮损自幼发生,沿左侧躯体呈带状分布,青春期进展迅速,组织病理见角化不全柱,符合线状汗孔角化症的诊断。该型被认为是体细胞发生突变成为携带突变基因的嵌合体,突变的体细胞与正常细胞交错镶嵌形成的皮肤嵌合体,致使皮损形成具有线状分布的特点,该型皮损具有最高的恶变风险。发生在面部的线状汗孔角化症时有报告,2005年Zhang等[10]报道了英国第1例局限于面部的先天性线状汗孔角化症。2016年 Basu等[4]报道了1例9岁女孩面部出现沿Blaschko线的线状汗孔角化症。文献报道汗孔角化症的恶变率为7.5%~11%,可发展为非黑素瘤皮肤癌,如鲍温病、基底细胞癌和鳞状细胞癌[11],尤其是播散性浅表性光化性汗孔角化症、巨大汗孔角化症和线状汗孔角化症,可能与遗传杂合性缺失有关[12]。
线状汗孔角化症临床上需与线状苔藓、线状扁平苔藓、线状银屑病、炎性线状疣状表皮痣和各种疣状色素失禁等疾病相鉴别,前者的板层状角化不全柱是主要鉴别要点。虽然角化不全柱对汗孔角化症的诊断是必要的,但并非特异性,这也可见于寻常疣、日光性角化病、脂溢性角化病等。本病治疗无特异性,基本原则是对症处理。液氮冷冻、CO2激光、光动力、手术切除等方法可以选用[13]。所有持续性存在的皮损均应长期随访警惕继发性皮肤癌,因此早期诊断和早期治疗非常重要。皮损面积广泛时可选择口服阿维A酯或异维A酸治疗,往往在用药期间有效,停药后可能复发。寻找致病基因,对该疾病进行产前诊断、基因治疗,是今后主要的研究方向。
本病例报告的特殊之处是罕见局限于左侧躯体的线状汗孔角化症,皮损沿Blaschko线分布,包括面部和生殖器均受累。由于其具有恶性肿瘤的风险,需要长期随访。该患者自幼发生的鼻腔异常和前额脂肪瘤是否与汗孔角化症相关目前尚不明确。
