鲜枸杞子浆质量标准研究*
2023-05-11齐喜红
田 雨,王 庆,齐喜红,韩 莎
(1. 宁夏医科大学药学院,宁夏 银川 750004; 2. 宁夏回族自治区药品检验研究院,宁夏 银川 750001)
枸杞子为药食同源的常用滋补类中药[1],始载于《神农本草经》[2],现收载于2020年版《中国药典(一部)》,具有滋补肝肾、益精明目功效[3]。枸杞子含有丰富的化学成分,主要有枸杞多糖、生物碱、氨基酸、黄酮类、维生素、微量元素[4-6]等。鲜枸杞子浆是结合现代科技,将枸杞子鲜果经破碎、制浆及灭菌工艺灌装生产而成,最大限度地保留了枸杞子鲜果的药效成分,同时便于运输、储存和使用。近年来,鲜枸杞子浆作为食品已有应用,其标准为食品行业团体标准[7-9]。为传承、创新枸杞子中药饮片传统炮制加工方法,提升其生物利用度,拓宽鲜枸杞子浆在中药领域的应用,本研究中采用薄层色谱(TLC)法对鲜枸杞子浆进行定性鉴别,分别采用紫外-可见分光光度法和高效液相色谱法测定枸杞多糖及甜菜碱的含量,为建立鲜枸杞子浆的质量标准提供参考。现报道如下。
1 仪器与试药
1.1 仪器
XS205型电子天平(瑞士梅特勒-托利多公司,精度为十万分之一);2550 型紫外分光光度计(日本岛津公司);安捷伦1260型高效液相色谱仪(美国Agilent公司)。
1.2 试药
枸杞子浆(宁夏早康生物科技有限公司,批号分别为20191001,20191002,20191003,20191004,20191005,20200701,20200702,20200703,20200704,20200705);枸杞子对照药材(批号为121072-201611),D-无水葡萄糖对照品(批号为110833-201908,纯度为99.8%),甜菜碱对照品(批号为110894 - 201604,纯度为99.2%),均购自中国食品药品检定研究院;乙腈为色谱纯,水为超纯水,其余试剂均为分析纯。
2 方法与结果
2.1 TLC 鉴别
取样品2.0 g,加水35 mL,加热煮沸15 min,放冷,滤过,滤液用乙酸乙酯15 mL 振摇提取,分取乙酸乙酯液,浓缩至1 mL,作为供试品溶液。取枸杞子对照药材0.5 g,按供试品溶液制备方法制备对照药材溶液。吸取供试品溶液和对照药材溶液各5 μL,用自动点样仪分别点在同一硅胶G薄层板上,以乙酸乙酯-三氯甲烷-甲酸(3∶2∶1,V/V/V)为展开剂,上行展开,展距8.0 cm,晾干,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液色谱相同位置显相同颜色的荧光斑点。色谱图见图1。
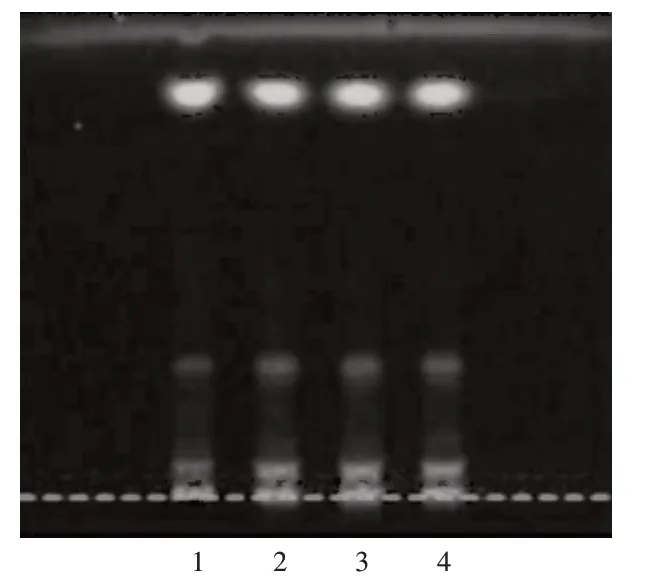
图1 薄层色谱图(365 nm)1.Reference medicinal materials solution 2-4.Test solutionFig.1 TLC chromatogram(365 nm)
2.2 枸杞多糖含量测定
2.2.1 溶液制备
取D-无水葡萄糖对照品25.11 mg,精密称定,置250 mL 容量瓶中,加水定容,即得对照品溶液。取样品2.0 g,精密称定,加乙醚100 mL,加热回流1 h,弃去乙醚液,残渣加100 mL 80%乙醇,加热回流提取1 h,滤过,用30 mL 80%乙醇分次洗涤,将滤渣和滤纸置装有150 mL 水的烧瓶中,继续加热回流提取2 h,趁热滤过,用少量热水冲洗滤器,收集滤液和洗液,放冷,转移至250 mL 容量瓶中,加水稀释并定容,摇匀,即得供试品溶液。
2.2.2 标准曲线制备[10-12]
精密量取2.2.1 项下对照品溶液0.2,0.4,0.6,0.8,1.0,1.5 mL,分别置具塞试管中,加水至2.0 mL,各精密加入5%苯酚溶液1 mL,摇匀,迅速精密加入硫酸5 mL,摇匀,放置10 min,40 ℃水浴放置15 min,取出,迅速冷却至室温,以溶剂为空白对照,于490 nm 波长处测定吸光度,以吸光度(Y)为纵坐标、质量浓度(X,μg/ mL)为横坐标绘制标准曲线,得线性方程Y=0.072X- 0.026 4(r= 0.999 8,n= 6)。结果表明,D-无水葡萄糖质量浓度在2.506 0~18.794 8 μg/ mL 范围内与吸光度线性关系良好。
2.2.3 样品含量测定方法
精密量取供试品溶液1.0 mL,置具塞试管中,精密加水1.0 mL,按2.2.2 项下自“各精密加入5%苯酚溶液1 mL”起,依法测定吸光度,按标准曲线计算供试品溶液中D-无水葡萄糖的浓度和含量。
2.2.4 方法学考察
精密度试验:精密量取D-无水葡萄糖对照品溶液0.4 mL,置具塞试管中,加水至2.0 mL,按2.2.3项下方法测定吸光度6 次。结果的RSD为0.11%(n= 6),表明仪器精密度良好。
重复性试验:取同一批(批号为20191001)样品6份,按2.2.1 项下方法制备供试品溶液,按2.2.3 项下方法测定吸光度,并计算含量。结果平均含量为0.81%,RSD为1.28%(n=6),表明方法重复性良好。
稳定性试验:取样品(批号为20191001),按2.2.1项下方法制备供试品溶液,分别于配制后0,2,4,6,8 h时按2.2.3项下方法测定吸光度。结果供试品溶液在室温下放置6 h的吸光度无明显变化,RSD为0.63%(n=4),表明供试品溶液在6 h 内稳定性良好;放置8 h 后吸光度下降,RSD为2.83%(n=5)。
加样回收试验:取已知含量的样品(批号为20191001)0.25 g,精密称定,共6 份,分别精密加入D-无水葡萄糖对照品2 mg,按2.2.1项下方法制备供试品溶液,按2.2.3项下方法进样测定,并计算加样回收率。结果见表1。

表1 枸杞多糖加样回收试验结果(n=6)Tab.1 Results of the recovery test of lycium burbarum polysaccharide(n=6)
2.2.5 样品含量测定
取10 批样品,按2.2.1 项下方法制备供试品溶液,按2.2.3项下方法进样测定,并计算含量。结果见表2。

表2 样品中枸杞多糖含量测定结果(%)Tab.2 Results of the content determination of lycium burbarum polysaccharide in samples(%)
2.3 甜菜碱含量测定
2.3.1 色谱条件
色谱柱:Merck Purospher@STAR NH2柱(250 mm ×4.6 mm,5 μm);流动相:乙腈- 水溶液(85∶15,V/V);流速:1.0 mL/min;检测波长:195 nm;柱温:25 ℃;进样量:10 μL。
2.3.2 溶液制备
取甜菜碱对照品17.68 mg,精密称定,置10 mL 容量瓶中,加水稀释并定容,摇匀,作为对照品溶液。取样品4 g,精密称定,置锥形瓶中,精密加入甲醇50 mL,称定质量,加热回流1 h,冷却,再次称定质量,用甲醇补足减失的质量,摇匀,滤过,精密量取续滤液2 mL,置碱性氧化铝固相萃取柱(2 g)上,加乙醇30 mL 洗脱,收集洗脱液,蒸干,残渣加水适量,溶解,转移至2 mL容量瓶中并定容,摇匀,滤过,取续滤液,即得供试品溶液[13-14]。同法制备阴性对照品溶液。
2.3.3 方法学考察
专属性试验:取2.3.2 项下对照品溶液、供试品溶液、阴性对照品溶液各适量,按2.3.1 项下色谱条件进样测定,记录色谱图。结果供试品溶液色谱中,在与对照品溶液色谱同一保留时间处有相应色谱峰,且阴性对照无干扰,表明方法专属性良好。色谱图见图2。
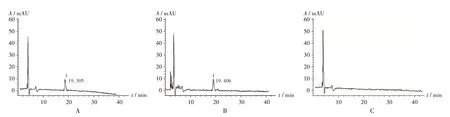
图2 高效液相色谱图1.BetaineA.Reference solution B.Test solution C.Negative reference solutionFig.2 HPLC chromatograms
线性关系考察:分别精密量取对照品溶液0.1,0.5,1.0,1.5,2.0 mL,置10 mL容量瓶中,加水定容,摇匀,按2.3.1项下色谱条件进样测定,以峰面积(Y)为纵坐标、质量浓度(X,μg/mL)为横坐标进行线性回归,得回归方程Y =1.952 7X -9.219 0(r= 0.999 6,n= 5)。结果表明,甜菜碱质量浓度在17.54~350.77 μg/ mL范围内与峰面积线性关系良好。
精密度试验:精密吸取2.3.2 项下对照品溶液,按2.3.1项下色谱条件进样测定6次,记录峰面积。结果的RSD为1.50%(n=6),表明仪器精密度良好。
重复性试验:取同一批(批号为20191001)样品6份,按2.3.2 项下方法制备供试品溶液,按2.3.1 项下色谱条件进样测定,记录峰面积,并计算含量。结果的RSD为1.10%(n=6),表明方法重复性良好。
稳定性试验:取2.3.2 项下对照品溶液,分别于0,2,4,6,8,12,24 h 时按2.3.1 项下色谱条件进样测定,记录峰面积。结果的RSD为1.94%(n=7),表明甜菜碱在24 h内稳定性良好。
加样回收试验:取已知含量的样品(批号为20191001)2 g,精密称定,共6 份,分别精密加入质量浓度为3.761 7 mg/ mL 的甜菜碱对照品溶液1 mL,按2.3.2 项下方法制备供试品溶液,按2.3.1 项下色谱条件进样测定,计算加样回收率。结果见表3。
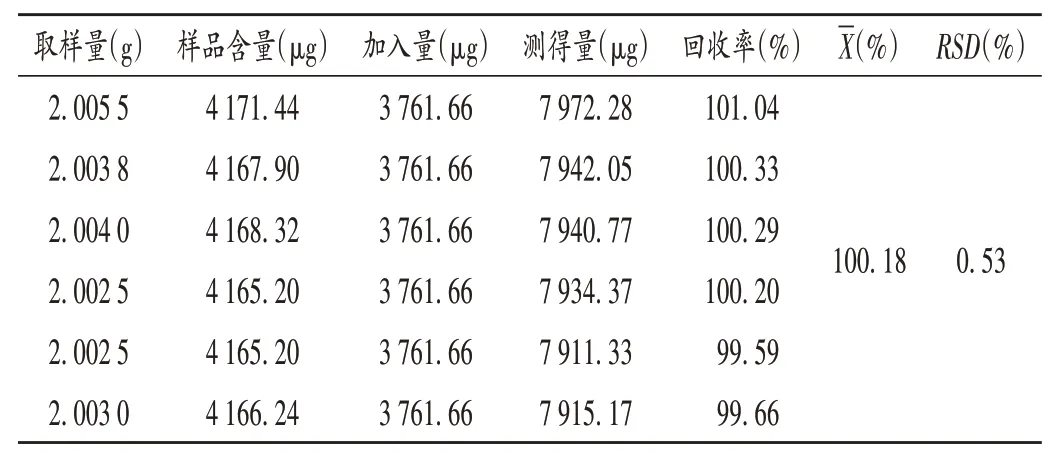
表3 甜菜碱加样回收试验结果(n=6)Tab.3 Results of the recovery test of betaine(n=6)
2.3.4 样品含量测定
取10 批样品,按2.3.2 项下方法制备供试品溶液,按2.3.1 项下色谱条件进样测定,记录峰面积,并计算含量。结果见表4。

表4 样品中甜菜碱含量测定结果(%)Tab.4 Results of the content determination of betaine in samples(%)
3 讨论
本研究中鲜枸杞子浆的TLC 鉴别主要参考2020年版《中国药典(一部)》枸杞子TLC鉴别项下的方法[3],同时考察了不同薄层板、不同温湿度对试验结果的影响,结果重复性良好,操作简便,斑点清晰,分离效果好。故确定为2.1项下鲜枸杞子浆TLC鉴别方法。
在190~680 nm 波长范围内扫描,结果于490 nm波长处葡萄糖吸收峰最大,故选用490 nm 为枸杞多糖的测定波长。考察了样品在不同提取时间(2,3,4 h)所测得的枸杞多糖含量,结果分别为0.82%,0.83%,0.84%。综合考虑,确定样品提取时间为2 h。考察了比色皿、波长和苯酚试剂的耐用性,结果均无明显影响。
从鲜果炮制成鲜枸杞子浆的收率为80%~90%,从鲜果炮制成枸杞子的收率为20%~25%,即鲜枸杞子浆与枸杞子干品比率为4∶1。参照2020年版《中国药典(一部)》枸杞子含量测定项下枸杞多糖的限度为不得少于1.8%,结合10 批样品的含量测定结果,暂将鲜枸杞子浆中枸杞多糖的限度订为不得少于0.5%。
考察了流动相乙腈- 水溶液的3 种比例(87∶13,85∶15,83∶17,V/V),结果均符合要求。考察了不同品牌色谱柱(Merck Purospher@STAR NH2柱、Agilent NH2柱、Waters NH2柱,规格均为250 mm×4.6 mm,5 μm)对甜菜碱含量测定结果的影响,结果对甜菜碱含量测定结果影响较小,故最终确定为2.3.1项下色谱条件。结合10批样品的甜菜碱含量测定结果,暂将鲜枸杞子浆中甜菜碱的限度订为不得少于0.10%。
综上所述,本研究中建立的方法简便、可行、重复性好,可用于鲜枸杞子浆的质量控制。
