分子排阻色谱-反相高效液相色谱法测定阿莫西林颗粒中高分子聚合物含量*
2023-05-11王显芹郑如程钱世兵傅世玲邢建辉
王显芹,郑如程,钱世兵,傅世玲,邢建辉,秦 亮
(1. 安徽安科恒益药业有限公司,安徽 铜陵 244061; 2. 安徽省铜陵市食品药品检验中心,安徽 铜陵 244061)
β-内酰胺类抗菌药物包括青霉素及其衍生物、头孢菌素、单酰胺环类、碳青霉烯类、青霉烯类抑制剂等,是临床常用的注射剂型抗菌药物[1]。阿莫西林是第2代青霉素类抗菌药物,具有广谱抗菌作用,为首选β- 内酰胺类口服抗菌药物[2],临床常用于治疗革兰阴性菌、革兰阳性菌等所致的感染性疾病、呼吸系统疾病及泌尿系统疾病[3-6],但药品不良反应发生率较高,过敏反应可引起β-内酰胺类抗菌药物聚合物杂质[7]。阿莫西林的主要致敏原为其二聚体、三聚体、四聚体和五聚体,聚合度越高,引发的过敏反应越强,故应严格控制阿莫西林中高分子聚合物的含量[8]。2020年版《中国药典(二部)》[9]中采用Sephadex-G10 葡聚糖凝胶柱对聚合物进行控制,但阿莫西林颗粒标准中未收录“聚合物”检查项。《美国药典(43 版)》[10]和《欧洲药典(9.0版)》[11]中采用反相高效液相色谱(RP-HPLC)法同时控制聚合物杂质和一般杂质。实际应用过程中,采用Sephadex-G10 葡聚糖凝胶柱分离测定聚合物时,存在长时间使用导致柱效降低,聚合物和主峰分离度不够,测定时间较长,测定过程烦琐,进样量大,对进样设备要求高等问题。本研究中参考文献[12-16],在国家药品监督管理局公布的参比制剂目录中选择国外L 公司的阿莫西林细粒(阿莫西林颗粒属改剂型品种,国外原研药无该剂型,相似剂型为细粒),以国内A公司生产的阿莫西林颗粒(参比制剂)为检测样本,建立了测定阿莫西林颗粒中高分子聚合物含量的分子排阻色谱-反相高效液相色谱法。现报道如下。
1 仪器与试药
1.1 仪器
SQP QUINTIX 224-1CN 型电子天平(德国赛多利斯集团,精度为0.1 mg/0.01 mg);S210 型pH 计(瑞士梅特勒-托利多集团);KQ-300DE型数控超声波清洗器(昆山市超声仪器有限公司,功率为300 W,频率为40 kHz);LC-20A型高效液相色谱仪(日本岛津公司),配有二极管阵列检测器。
1.2 试药
阿莫西林颗粒(A 公司,批号分别为S190601,S190801,S200301,S200302,S200303);阿莫西林细粒(L 公司,批号分别为1903A0,9420);阿莫西林对照品(中国食品药品检定研究院,批号为130409 - 201512,含量为86.9%);乙腈为色谱纯,其余试剂均为分析纯,试验用水由Milli-Q系统制备。
2 方法与结果
2.1 色谱条件
色谱柱:Cef-SEC球状亲水硅胶柱(300 mm×7.8 mm,5 μm);流动相:磷酸盐缓冲液[pH 7.0,0.005 mol/L 磷酸氢二钠-0.005 mol/L 磷酸二氢钠(50∶50,V/V)]-乙腈(90∶10,V/V);流速:0.6 mL / min;检测波长:254 nm;柱温:25 ℃;进样量:20 μL。
2.2 溶液制备
供试品溶液:取样品适量,精密称定,冷水中加流动相适量,超声5 min使溶解,加流动相定量稀释成每1 mL含阿莫西林1.0 mg的溶液,滤过,取续滤液,即得。
对照品溶液:取阿莫西林对照品适量,精密称定,加流动相溶解,稀释成每1 mL含阿莫西林10 μg的溶液,即得。
系统适用性溶液:取阿莫西林对照品10 mg,精密称定,置10 mL 容量瓶中,加流动相5 mL,超声使溶解,加0.1 mol/L 氢氧化钠溶液1 mL,30 ℃水浴10 min,用0.1 mol/L 盐酸溶液调酸碱性至中性,加流动相定容,摇匀,即得。
2.3 方法学考察
系统适用性试验:分别取供试品溶液、对照品溶液、系统适用性溶液各适量,按2.1项下色谱条件进样测定。结果系统适用性溶液色谱图中,阿莫西林峰与主要降解杂质峰的分离度为7.8,符合要求。色谱图见图1。
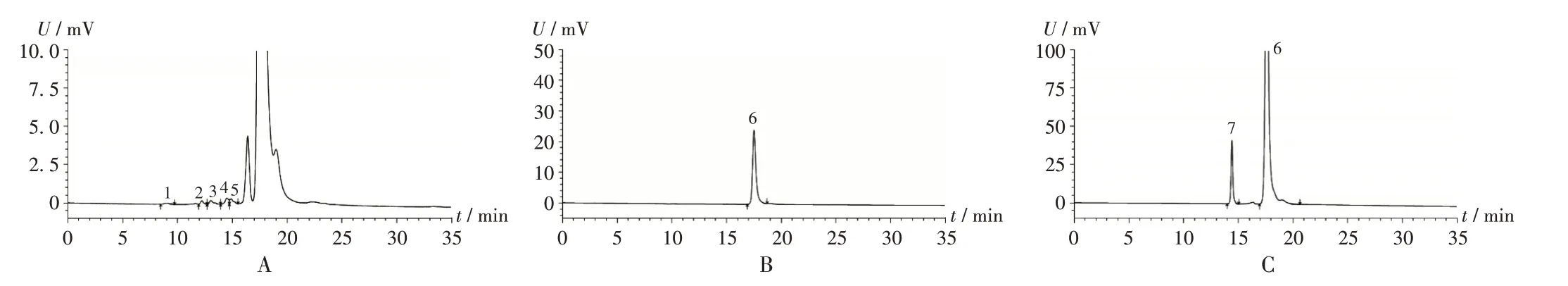
图1 系统适用性试验高效液相色谱图1-5.Polymer impurities 6.Amoxicillin 7.Main degradation impurities A.Test solution B.Reference solution C.System suitability solutionFig.1 HPLC chromatograms of the system suitability test
专属性试验:由药品说明书可知,样品中含有蔗糖、日落黄铝色淀、糊精、枸橼酸钠、香兰素、桔子香精粉末等辅料。按处方工艺分别制备缺主药的单一辅料溶液及混合辅料溶液,按2.1 项下色谱条件进样测定。结果显示,辅料中存在干扰峰(蔗糖峰和日落黄铝色淀峰),故测定聚合物含量时需扣除。色谱图见图2。
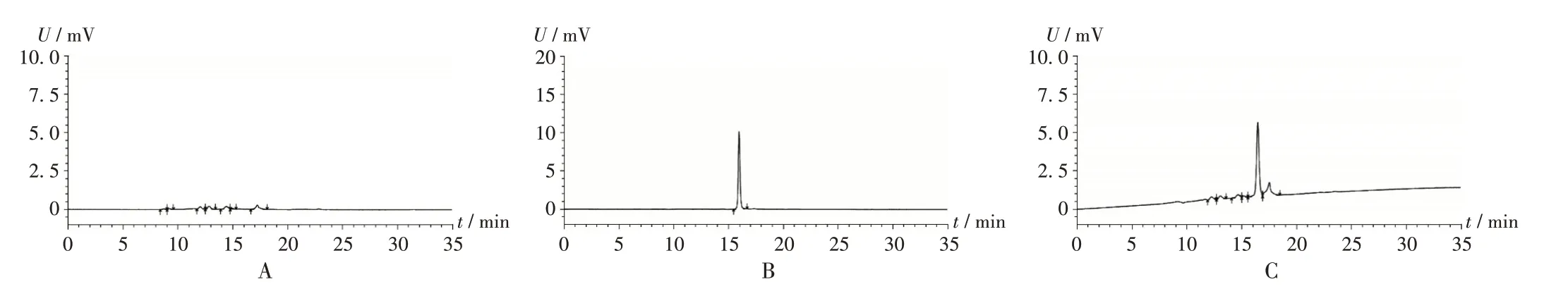
图2 专属性试验高效液相色谱图A.Sucrose solution B.Sunset yellow aluminum lake solution C.Mixed excipient solutionFig.2 HPLC chromatograms of the specific test
破坏性试验:取样品和混合辅料,分别进行碱破坏(加0.1 mol/L 氢氧化钠溶液0.8 mL,室温放置5 min,用0.1 mol/L 盐酸溶液调至中性)和高温破坏(80 ℃破坏8 h)后,配制成相应的强制降解供试品溶液和混合辅料溶液,按2.1项下色谱条件进样测定,记录色谱图。由图3 可知,辅料中存在干扰峰;经碱破坏和高温破坏的混合辅料对阿莫西林中聚合物的测定未产生明显干扰,阿莫西林峰与其前主要降解杂质峰间的分离度均大于1.5,符合要求。

图3 破坏性试验高效液相色谱图1.AmoxicillinA1,A2.Test solution(alkali damage and high-temperature damage) B1,B2.Mixed excipient solution(alkali damage and high-temperature damage)Fig.3 HPLC chromatograms of the destroyed test
线性关系考察:取阿莫西林对照品28.83 mg,精密称定,加流动相配制成质量浓度为50.1 μg/mL 的线性贮备液;分别精密量取0.5,1.0,2.0,3.0,4.0,5.0,6.0,7.0 mL,置10 mL 容量瓶中,加流动相定容,摇匀,即得质量浓度分别为2.51,5.01,10.02,15.03,20.04,25.05,30.06,35.07 μg/mL 的系列线性溶液。按2.1项下色谱条件进样测定,以峰面积(A)为纵坐标、质量浓度(C,μg/mL)为横坐标进行线性回归,得回归方程A= 6 278.3C- 2 554.0(r=0.999 3,n=8)。结果表明,阿莫西林对照品溶液质量浓度在2.51~35.07 μg/mL范围内与峰面积线性关系良好。
检测限和定量限确定:精密量取线性关系考察项下质量浓度为2.51 μg/ mL 的线性溶液,逐级稀释,按2.1项下色谱条件进样测定,记录色谱图。取信噪比(S/N)分别为10∶1 和3∶1 时待测成分的质量为定量限和检测限,结果分别为5.00 ng和1.60 ng。
精密度试验:取线性关系考察项下质量浓度为10.02 μg/mL的线性溶液,按2.1项下色谱条件进样测定6 次,记录色谱图。结果峰面积的RSD为0.60%(n=6),表明仪器精密度良好。
稳定性试验:按2.2项下方法制备对照品溶液1份,于室温下放置0,2,4,6,8 h时按2.1项下色谱条件进样测定,以峰面积变化考察对照品溶液的稳定性。结果对照品溶液主峰峰面积的RSD为0.5%(n=5),表明对照品溶液室温放置8 h 内稳定性良好。按2.2 项下方法制备供试品溶液2 份,取1 份供试品溶液于室温下放置0,1,2,3,4,5,6,8 h 及另1 份供试品溶液于4 ℃下放置0,2,4,6,8 h 时按2.1 项下色谱条件进样测定,按外标法以峰面积计算聚合物含量,以聚合物含量变化量考察供试品溶液的稳定性。与0 h 相比,供试品溶液室温、4 ℃条件下放置8 h 聚合物含量分别增加0.40%和0.04%,表明供试品溶液在室温条件下不稳定,需临用新制,或应保存于4 ℃下并在8 h内使用。
重复性试验:取同一批(批号为S190601)样品,按2.2 项下方法制备供试品溶液6 份,按2.1 项下色谱条件进样测定,按外标法以峰面积计算聚合物含量。结果6 份供试品溶液中聚合物含量分别为0.33%,0.28%,0.26%,0.26%,0.26%,0.25%,聚合物含量最大变化量为0.08%,表明方法重复性良好。
中间精密度试验:由不同分析人员按重复性试验项下方法,在不同时间重新制备6 份供试品溶液。结果聚合物含量分别为0.30%,0.28%,0.27%,0.32%,0.27%,0.26%。12份供试品溶液中,聚合物含量最大变化量为0.08%,表明方法中间精密度良好。
耐用性试验:在其他条件不变的情况下,分别微调流速、柱温、磷酸盐缓冲液pH、流动相比例、检测波长,分别配制系统适用性溶液、对照品溶液和供试品溶液,测定聚合物含量。结果测定的聚合物含量最大变化量为0.08%,考虑聚合物含量为多个聚合物峰及积分差异,测定误差均在可接受范围内;系统适用性溶液中,主峰与较大杂质峰间的分离度均大于1.5。结果见表1,由于蔗糖色谱图中存在多个小的色谱峰,无法用保留时间描述,故未列出。

表1 耐用性试验考察结果Tab.1 Results of the durability test
2.4 样品含量测定
取5 批A 公司生产的阿莫西林颗粒,以2 批次L 公司生产的阿莫西林细粒为参比制剂,按2.1项下色谱条件进样测定,按外标法以峰面积计算聚合物含量。结果A 公司5 批次阿莫西林颗粒中高分子聚合物的含量均低于参比制剂,提示A公司阿莫西林颗粒的质量不低于参比制剂。详见表2。

表2 样品中高分子聚合物含量测定结果(%)Tab.2 Results of the content determination of high - molecular polymer in samples(%)
3 讨论
3.1 柱温选择
参考文献[14]中的色谱条件(柱温为30 ℃,其余色谱条件不变)对各单一辅料溶液、混合辅料溶液及供试品溶液进行测定。结果糊精、枸橼酸钠、香兰素及桔子香精粉末对聚合物检测基本无干扰;蔗糖与聚合物出峰位置有重合,故在计算时需扣除蔗糖峰中保留时间与聚合物峰重合的峰面积;日落黄铝色淀峰于主峰前出峰,但峰形较差,故调整柱温为25 ℃,此温度下的峰形更优。
3.2 超声时间选择
取同一批(批号为S190601)样品4 份,按2.2 项下方法制备供试品溶液,样品溶解时分别超声处理3,5,7,10 min,按2.1 项下色谱条件进样测定,按外标法以峰面积计算阿莫西林颗粒中聚合物含量。结果超声3 min 与超声10 min 的聚合物含量基本不变,为保证样品溶解完全及检测结果的可重复性,同时避免超声时间过长产生的热量导致聚合物含量发生变化,建议超声时间为5 min。
3.3 溶解时水温选择
根据阿莫西林颗粒药品说明书中的临床用法,使用前需用凉开水搅拌后服用。结合本品实际使用情况(即每次1 袋),取A 公司生产的阿莫西林颗粒,制备含质量浓度为3.75 mg/mL 的供试品溶液,溶解时的水温分别选择(25 ± 2)℃(凉开水)、40~50 ℃(温水)、70~80 ℃(热水),于室温下放置3 h,考察0,1,2,3 h 时聚合物的含量。结果聚合物的含量随溶解时水温的升高而增加,聚合物含量越高可能导致的过敏反应发生率越高。故为保证测定结果的准确性和可重复性,建议样品于冷水中用流动相溶解。
3.4 聚合物限度确定
根据2020年版《中国药典(四部)》附录[17]中有关的指导原则和ICH HARMONISED TRIPARTITE GUIDELINE IMPURITIES IN NEW DRUG PRODUCTS Q3B(R2)中对杂质限度的规定,同时结合本研究中的稳定性试验结果及参比制剂的对比结果,将本品的聚合物限度拟订为≤标示量的3.0%。
3.5 方法评价
本研究中所建立的方法操作简便、专属性强、结果准确、重复性好,可用于阿莫西林颗粒中高分子聚合物的含量测定。
