一种氧杂蒽酮类海洋抗污损天然产物振动光谱的理论研究
2023-05-08梁小蕊张纪磊孙晓伟
梁小蕊,李 荫,张纪磊,柳 叶,孙晓伟
(海军航空大学,山东烟台 264001)
海洋生物污损是指海洋生物(包括海洋植物、动物和海洋微生物)在船底、码头、浮标及各类人工设施上附着、栖息及生长并对人类活动产生不利影响的现象和过程,这些附着生物被称为污损生物[1-2]。海洋生物污损给水产养殖、国防、航运及海洋工程等带来了多方面的严重危害。例如:航运船只被污损生物附着后会增加航行阻力,堵塞管道,加快金属被腐蚀的速度;污损生物还会妨碍和干扰海中军事设施和民用器材的正常工作,改变大型海洋结构物水下部位的表面特征等[3-4]。因此,研究具有抗污损活性的化合物,并将其应用于舰船等海洋设施中,具有重要意义。
大量研究结果表明,海洋真菌能够产生结构新颖、高效及环境友好型的抗污损活性化合物。因此,对于海洋真菌次生代谢产物的研究成为了近年来的热点[5-9]。对于次生代谢产物研究的1个重要方面就是单体化合物的结构鉴定,目前采用的主要方法是由核磁共振波谱、红外光谱、拉曼光谱及质谱等波谱分析方法相互配合进行结构鉴定。其中,红外光谱、自然拉曼光谱及核磁共振波谱,已成为鉴定各种有机、无机化合物结构的有力工具,但其谱峰较为复杂,不易分辨,而通过理论计算将光谱数据与实验数据进行对比分析,则会大大提高鉴定效率[10-14]。
本文利用Gaussian 09W软件包,采用密度泛函理论(Density functional theroy,DFT)方法,对来自红树林未知真菌ZSUH-36 的氧杂蒽酮类化合物6,8-di-Omethyl versiconol 进行了理论研究,该化合物对B.amphitrite金星幼虫表现出中等强度的抗污损活性,其半数抑制浓度EC50值为5.30 μg/mL,即该化合物能够抑制金星幼虫的附着,可以作为有效的抗污损先导化合物[15]用于海洋生物污损的治理。本文采用DFTB3LYP理论方法计算了该化合物的最稳定构型,在该结构的基础上,计算了该化合物分子的红外振动频率和拉曼光谱振动频率;然后,对理论计算结果进行分析,探讨谱峰的归属,为海洋抗污损天然产物的结构鉴定提供了理论依据。
1 理论计算
DFT是1种研究多电子体系电子结构的量子力学方法。其显著的优势就是在不明显增加计算量的同时,还能考虑到电子相关,因而能够直观反映分子的振动信息[16-18]。6,8-di-O-methyl versiconol分子主要由C、H、O 等元素构成,杂化密度泛函B3LYP 方法在轻元素构成的分子计算中被广泛应用,因而,本文采用DFT 和B3LYP 方法,在3-21G 和6-311G 基组水平上,针对6,8-di-O-methyl versiconol 分子进行几何结构全优化。在分子最稳定构型的基础上,采用同样的理论方法,计算6,8-di-O-methyl versiconol 的红外振动光谱和拉曼光谱,全部计算均利用Gaussian 09W软件包完成。
2 结果与讨论
2.1 分子的空间几何构型
利用ChemOffice 绘制6,8-di-O-methyl versiconol分子的平面结构,如图1所示。
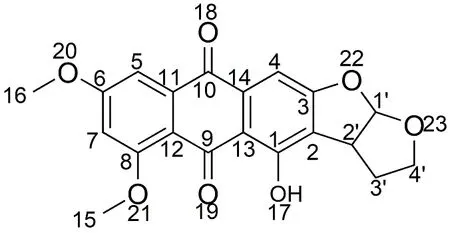
图1 6,8-di-O-methyl versiconol分子平面结构Fig.1 Planar structure of 6,8-di-O-methyl versiconol
量子化学计算过程中:首先,用Gaussian view6.0对6,8-di-O-methyl versiconol 分子的空间立体构型进行建模;再基于Gaussian 09W 软件包,采用DFTB3LYP 理论方法,选择3-21G 基组对分子构型进行粗优化,在该结构的基础上,选择6-311G 基组进行结构的再优化,得到分子的最稳定三维立体构型。计算结果没有虚频,说明结构合理。将优化后的分子立体构型以及笛卡尔坐标列于图2 中,分子中的原子编号参照图1。
优化后的6,8-di-O-methyl versiconol 分子为三维非平面结构,如图2所示。
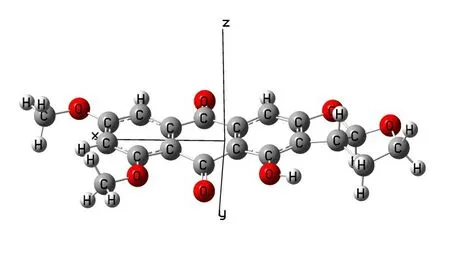
图2 优化后的6,8-di-O-methyl versiconol分子结构Fig.2 Optimized structure of 6,8-di-O-methyl versiconol
分子结构以9,10-蒽二酮环为主体:2C和3C上连了2个四氢呋喃五元环;1C上连了1个羟基;6C、8C上分别连了1 个甲氧基。将优化后分子的键长、键角和二面角数据列于表1。
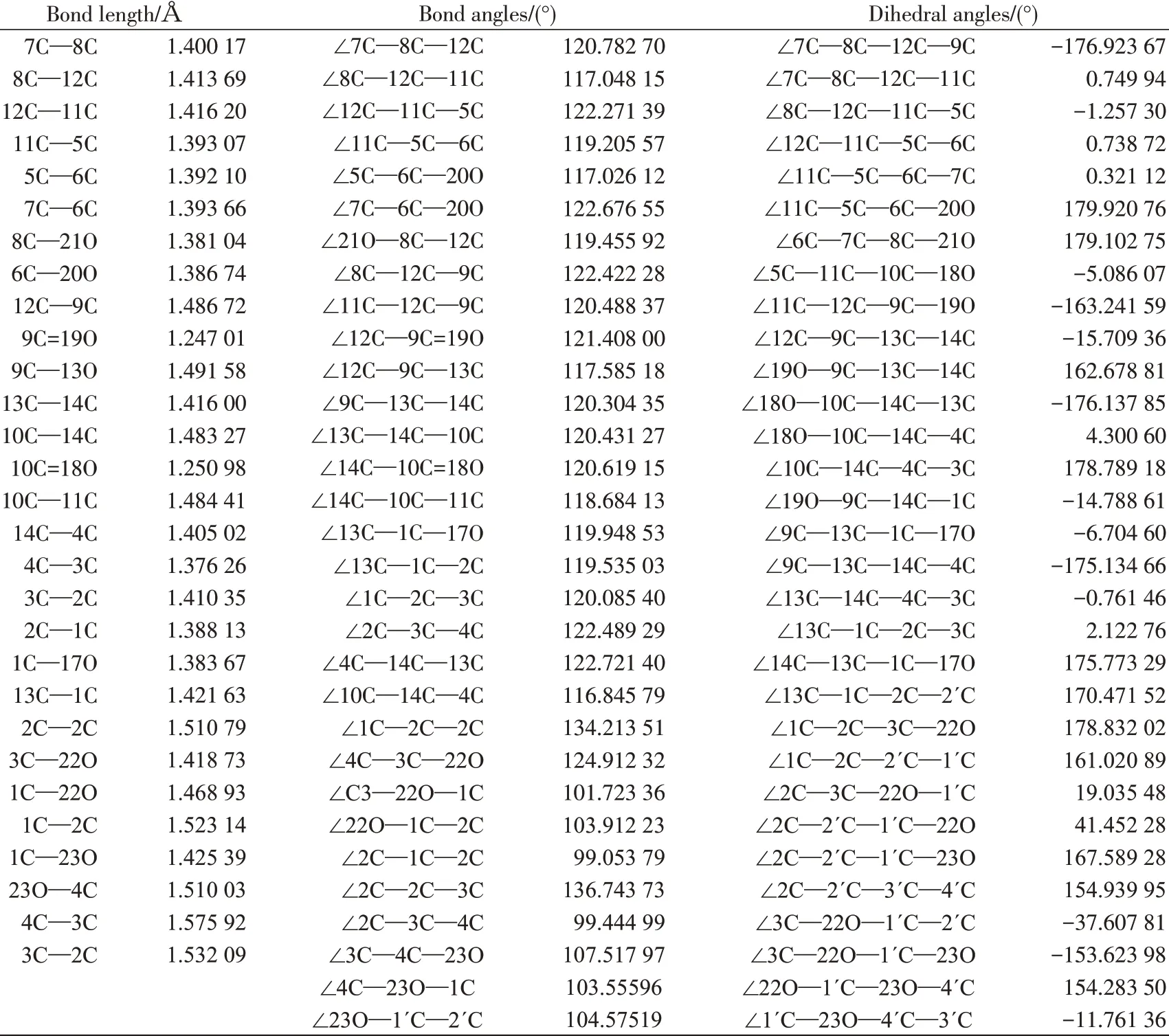
表1 优化后6,8-di-O-methyl versiconol分子的键长、键角和二面角数据Tab.1 Optimized bond lengths,bond angles and dihedral angles of 6,8-di-O-methyl versiconol
由表1中的二面角数据可知,9,10-蒽二酮环主体结构中∠7C—8C—12C—11C、∠8C—12C—11C—5C、∠12C—9C—13C—14C、∠13C—1C—2C—3C 等分别为0.749 94°、-1.257 30°、-15.709 36°和2.122 76° ,这说明9,10-蒽二酮环主体结构的3 个六元环均不在同一平面,其中,两侧的苯环扭曲角度较小,而中间的二酮六元环扭曲角度较大。∠9C—13C—1C—17O、∠11C—5C—6C—20O、∠6C—7C—8C—21O 分别为-6.704 60°、179.920 76°、179.102 75°,这些数据说明1个羟基和2 个甲氧基与9,10-蒽二酮环主体之间也有一定的夹角。
从表1中的键长数据来看,9,10-蒽二酮环主体中间的二酮六元环中各个碳碳键长大多比未取代时的键长要短,而两侧苯环的各个碳碳键长大多比未取代时的键长略长,这也证实了上述的非平面结构,并且这些键长的变化说明9,10-蒽二酮环在分子中已形成了1个大的π 共轭体系。
2.2 前线分子轨道分析
采用DFT-B3LYP 理论方法:首先,用3-21G 基组对6,8-di-O-methyl versiconol 分子进行结构粗优化,优化后分子能量为-1 219.58 Hartree,单位换算后为-33 003.77 eV;在粗优化结构的基础上,用6-311G 基组 进 行 再 优 化 ,结 果 分 子 的 能 量 为-1 219.58 Hartree,换算后为-33 184.82 eV。可见,优化后的分子能量很低,且二次优化后的能量更低,这说明了优化构型满足最低能量状态。利用同样的理论方法,对分子的前线轨道进行了计算,进而得到了6,8-di-O-methyl versiconol 分子的最高占有轨道HOMO和最低空轨道LUMO的电子云分布图,如图3所示。
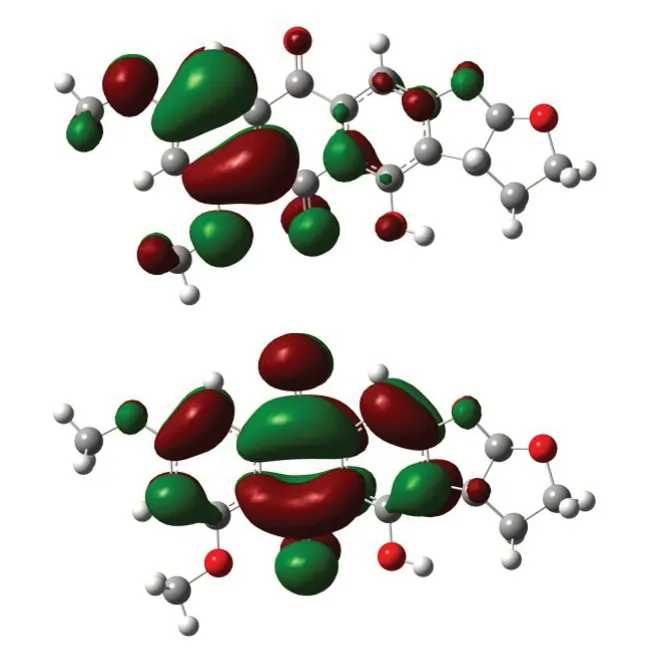
图3 6,8-di-O-methyl versiconol分子的HOMO(上)和LUMO(下)图Fig.3 Highest Occupied Molecular Orbital(above)and Lowest Unoccupied Molecular Orbital(following)of 6,8-di-O-methyl versiconol
从图3 可见6,8-di-O-methyl versiconol 分子中存在离域大π 键,从计算后HOMO和LUMO能量来看,其能量分别为-8.86 eV 和-6.62 eV,能隙为2.24 eV,能隙较小,说明该分子的离域π 电子容易被激发。
2.3 6,8-di-O-methyl versiconol 分子红外光谱分析
红外光谱可以用来对化合物进行定性鉴定、定量分析和结构分析,这些分析都离不开谱图的解析[19]。本文利用DFT-B3LYP 理论方法在前述优化构型的基础上,选择6-311G 基组对6,8-di-O-methyl versiconol分子在400~4 000 cm-1范围内的红外振动频率进行计算,利用Gaussian view6.0软件绘制了分子的理论红外振动光谱(如图4 所示),并根据红外光谱图上出现的吸收带位置、强度等信息对谱图进行了解析。
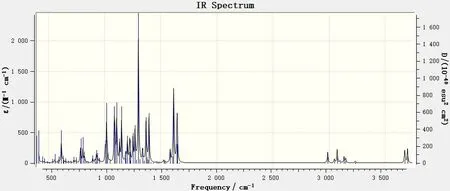
图4 400~4 000 cm-1范围内的6,8-di-O-methyl versiconol分子的理论红外谱图Fig.4 DFT IR spectrum of 6,8-di-O-methyl versiconol in the 400~4 000 cm-1 range
由图4可见,6,8-di-O-methyl versiconol分子的红外吸收峰数量较多,且强度明显。为了更清楚地辨别各个吸收峰的位置,按照红外吸收峰的特点,将分子的红外谱图分成3 个区域:400~1 000 cm-1(见图5),1 000~1 700 cm-1(见图6)和2 900~3 800 cm-1(见图7)。
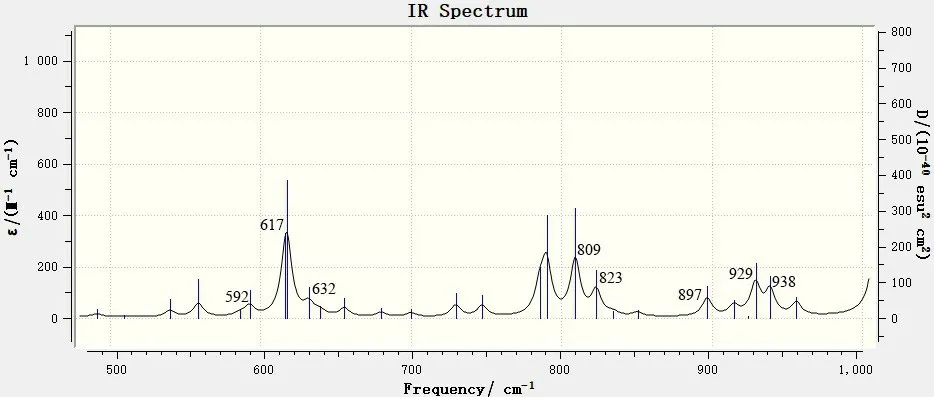
图5 400~1 000 cm-1范围内的6,8-di-O-methyl versiconol分子的理论红外谱图Fig.5 DFT IR Spectrum of 6,8-di-O-methyl versiconol in the 400~1 000 cm-1 range
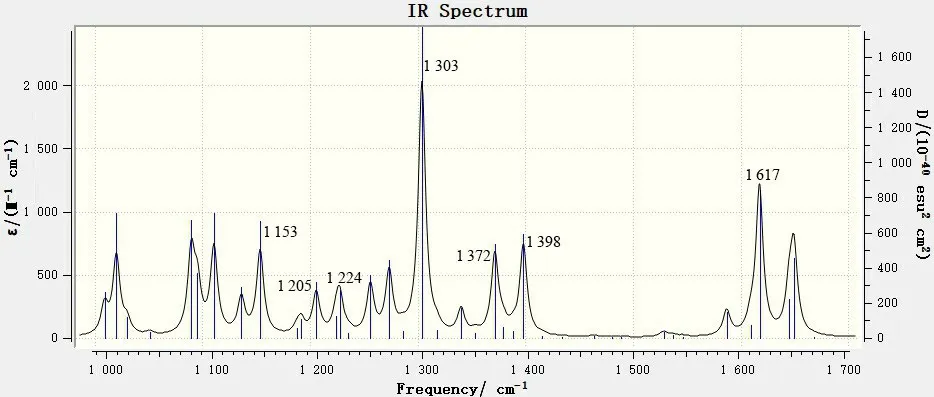
图6 1 000~1 700 cm-1范围内的6,8-di-O-methyl versiconol分子的理论红外谱图Fig.6 DFT IR spectrum of 6,8-di-O-methyl versiconol in the 1 000~1 700 cm-1 range
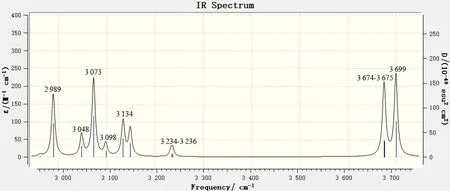
图7 2 900~3 800 cm-1范围内的6,8-di-O-methyl versiconol分子的理论红外谱图Fig.7 DFT IR spectrum of 6,8-di-O-methyl versiconol in the 2 900~3 800 cm-1 range
分析图5 的振动频率及吸收峰可知:400~1 000 cm-1范围内的红外吸收峰主要是由分子中各环的变形振动引起的,如592、617、632 cm-1处的吸收峰源于9,10-蒽二酮环的变形振动,这个区域的最强吸收峰出现在617 cm-1处;在809 cm-1处的吸收峰源于苯环上未被取代的7C—H 的弯曲振动;823、897、929 cm-1处的吸收峰主要是4C所在五元环上的C—H不对称变形振动引起的;938 cm-1处的吸收峰源于5C—H、4C—H的面外变形振动。
由图4 和图6 可知,1 000~1 700 cm-1频率范围内出现的红外吸收峰数量最多,峰强也最强。其中:最强吸收峰在1 303 cm-1处,是由9,10-蒽二酮环的面内变形振动及羟基和甲氧基的面内摇摆振动引起的;次强吸收峰在1 617 cm-1处,峰的形状较尖锐,可以归属为9,10-蒽二酮环中的羰基伸缩振动,在羰基化合物的红外谱图中,羰基的吸收一般为最强峰或次强峰;1 645~1 649 cm-1处的稍宽形状的峰为苯环的骨架伸缩振动峰。其他较强的特征峰包括:1 089~1 095 cm-1处的宽峰为五元环上C—H不对称变形振动峰;1 153、1 205和1 224 cm-1处的吸收峰为羟基和甲氧基的平面摇摆振动峰;1 372、1 398 cm-1处的2个强峰归属为苯环面内变形振动和羟基的平面摇摆振动峰。
图7中,2 900~3 800 cm-1区域内出现的振动吸收峰主要是伸缩振动频率,这个区域是整个光谱图中吸收最弱的区域。其中:3 674~3 699 cm-1处的中等强度吸收峰由是分子中羟基17O—H 的伸缩振动引起的;3 234~3 236 cm-1处的吸收峰是该分子中苯环上的3个碳氢键的伸缩振动峰,归属为4C—H、5C—H 和7C—H;3 134 cm-1处的吸收峰为3C 和4C 上的4 个碳氢键的不对称伸缩振动,而3 048 cm-1处的吸收峰则为3C和4C 上的4 个碳氢键的对称伸缩振动引起的;3 073 cm-1处的吸收峰归属为2个甲氧基的伸缩振动;2 989 cm-1处的吸收峰归属为1C—H的伸缩振动峰。
2.4 6,8-di-O-methyl versiconol 分子的理论拉曼光谱分析
拉曼光谱对于有机化合物的结构变化比红外光谱更敏感:化合物分子中有些无红外活性的振动,在拉曼光谱中有活性振动;有些在红外光谱中吸收较弱的官能团,在拉曼光谱中则呈现较强的振动峰。2 种光谱能够起到互补的作用,协同使用能够更加准确地鉴定化合物的结构。本文在6,8-di-O-methyl versiconol 最优构型的基础上,采用同样的理论方法和基组,计算了6,8-di-O-methyl versiconol 分子的理论拉曼光谱,如图8所示。
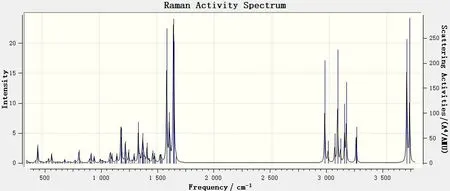
图8 6,8-di-O-methyl versiconol在400~4 000 cm-1范围内的理论拉曼光谱Fig.8 DFT Raman of 6,8-di-O-methyl versiconol molecule in the 400~4 000 cm-1 range
同样,为了便于分析,根据拉曼活性振动的情况,现将拉曼光谱分为 400~1 000 cm-1(图9)、1 000~1 700 cm-1(图10)、2 900~3 800 cm-1(图11)3 个部分进行讨论。
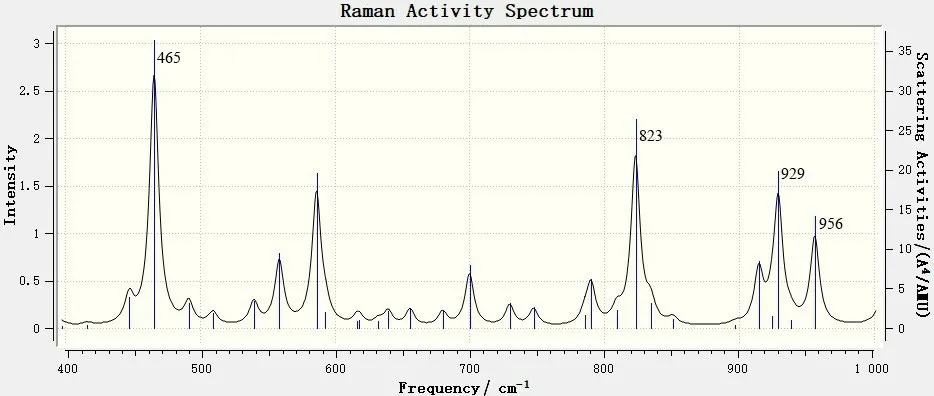
图9 6,8-di-O-methyl versiconol在400~1 000cm-1范围内的理论拉曼光谱Fig.9 DFT Raman of 6,8-di-O-methyl versiconol molecule in the 400~1 000 cm-1 range
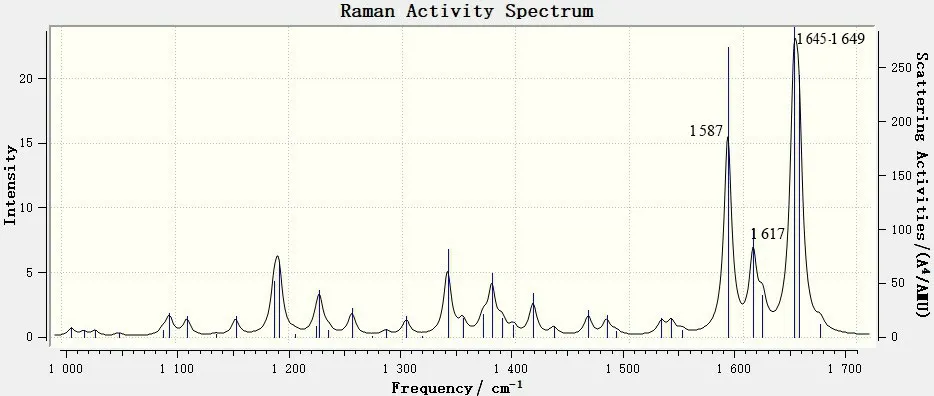
图10 6,8-di-O-methyl versiconol在1 000~1 700cm-1范围内的理论拉曼光谱Fig.10 DFT Raman of 6,8-di-O-methyl versiconol molecule in the 1 000~1 700cm-1 range.
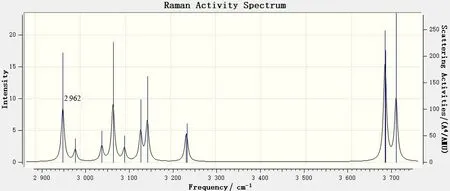
图11 6,8-di-O-methyl versiconol在2 900~3 800cm-1范围内的理论拉曼光谱Fig.11 DFT Raman of 6,8-di-O-methyl versiconol in the 2 900~3 800 cm-1 range
由图8 可知,6,8-di-O-methyl versiconol 分子在400~1 000 cm-1范围内的特征峰在3 个区域内是最弱的。从图9 可见:这个区域中相对最强的峰出现在465cm-1处,这是由蒽酮中C—C=O基的面内弯曲振动引起的,这一特征峰在红外光谱中吸收非常弱;而在红外光谱中有明显吸收的592、617、632 cm-1这3 处峰,在拉曼光谱中则没有体现;拉曼光谱中823、929和956 cm-1这3处的峰与上述红外光谱的吸收峰一致。
对照图4 和图8 可见,1 000~1 700cm-1区域内的拉曼谱峰与红外谱峰相似,都是中红外区中特征峰最多、最强的1 个区域,但二者也有明显的区别:有些在红外谱图中很强的峰,在拉曼谱图中则较弱,如红外光谱中1 303 cm-1处的最强吸收峰,1 089~1 095 cm-1处较强的宽峰,1 153、1 205 和1 224 cm-1处的羟基和甲氧基平面摇摆振动峰,以及1 372、1 398 cm-1处的2个强峰,在拉曼光谱中均显示为极弱的振动峰;而有一些在红外光谱中较弱的吸收峰,在拉曼光谱中则较强,如1 587 cm-1处的9,10-蒽二酮环的对称伸缩振动峰在拉曼光谱中为较强振动峰,而在红外光谱中则较弱;另外,拉曼光谱中 1 617 cm-1处以及1 645~1 649 cm-1处的稍宽形状的峰与红外光谱中的吸收峰位置和强度均相当。
比较图4 和图8 可见,在红外光谱中2 900~3 800 cm-1范围内的吸收峰最弱,而在拉曼光谱中,此范围内的振动峰则是非常强的。从图7 和图11可见,此区域内,从2 989 cm-1到3 699 cm-1处,共9 个振动峰,其峰位、振动形式均与红外光谱图中的一致。图11中,2 962 cm-1处有一非常强的拉曼振动峰,在红外光谱图中是没有的,这是6,8-di-O-methyl versiconol 分子中连接2 个五元环的2C—H 和1C—H 键的伸缩振动引起的。
3 结论
本文基于Gaussian 09W软件包,采用密度泛函理论中的杂化密度泛函B3LYP 方法,首先,用较低基组3-21G 对氧杂蒽酮类化合物6,8-di-O-methyl versiconol的分子结构进行了粗优化,在粗优化结构的基础上又用6-311G基组对分子结构进行了再优化,得到了分子的最稳定构型,计算结果无虚频,证明结果可靠。对6,8-di-O-methyl versiconol 最稳定构型进行分析,结果显示,以9,10-蒽二酮环为主体的分子是1 个三维非平面的结构,并且分子内部形成了1 个大的π共轭体系,对分子的稳定性更有利。以优化后的最稳定构型为基础,采用B3LYP/6-311G方法基组,计算了6,8-di-O-methyl versiconol 分子的理论红外光谱和拉曼光谱,分析探讨了2种光谱的特征峰的位置、振动特点以及振动模式等。
分析结果显示,红外光谱中2 900~3 800 cm-1范围内的吸收峰最弱,而在拉曼光谱中,此范围内的振动峰则是非常强的,这主要是由伸缩振动引起的。其中2 962 cm-1处的强拉曼振动峰,在红外光谱图中没有被吸收,这是6,8-di-O-methyl versiconol分子中连接2个五元环的2C—H 和1C—H 键的伸缩振动引起的。1 000~1 700 cm-1区域内的红外光谱与拉曼光谱相似,都是中红外区中特征峰最多、最强的1个区域,但二者也有较明显的区别,有些在红外谱图中很强的峰,在拉曼谱图中则较弱,400~1 000 cm-1范围内的特征峰在拉曼光谱中是最弱的,而在红外光谱中则稍强。从上述研究结果可知,将红外和拉曼2 种光谱结合进行分析可获得关于6,8-di-O-methyl versiconol 分子结构的丰富完整的信息。综上所述,本文的研究结果可为氧杂蒽酮类海洋抗污损天然产物分子的振动光谱检测和分子结构的鉴定提供光谱解析方面的理论依据。
