Alterations in the Gut Microbiome in Liver Recipients with Post-transplant Diabetes Mellitus
2023-03-22QiLingYuqiuHanYueMaXiaosenWangZhengZhuJingyuWangJiayingCaoLinXiaohanJunWangBaohongWang
Qi Ling, Yuqiu Han, Yue Ma, Xiaosen Wang, Zheng Zhu, Jingyu Wang, Jiaying Cao,Lin Xiaohan, Jun Wang*, Baohong Wang,h,*
a State Key Laboratory for Diagnosis and Treatment of Infectious Diseases,National Clinical Research Center for Infectious Diseases,Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
b Jinan Microecological Biomedicine Shandong Laboratory, Jinan 250117, China
c Department of Surgery, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
d Key Lab of Combined Multi-Organ Transplantation, Ministry of Public Health, Hangzhou 310003, China
e CAS Key Laboratory for Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China
f University of Chinese Academy of Sciences, Beijing 100101, China
g Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan 250117, China
h Research Units of Infectious Disease and Microecology, Chinese Academy of Medical Sciences, Hangzhou 310003, China
Keywords:Post-transplant diabetes mellitus Tacrolimus Metagenomics Metabolomics
ABSTRACT Post-transplant diabetes mellitus (PTDM) increases the risk of graft failure and death in liver transplant(LT)recipients.Experimental studies have indicated that enteric dysbiosis mediated by immunosuppressive tacrolimus(TAC)could contribute to glucose disorders,but no data on human recipients with PTDM have been reported.Here, by combining high-throughput shotgun metagenomics sequencing and metabolomics profiling, we characterized the intestinal microbiome (IM) in LT recipient cohort with or without PTDM and deciphered the potential relationship among IM,TAC dosage, and diabetes.By comparing with both non-PTDM and classical type 2 diabetes mellitus(T2DM),we identified microbial signatures of PTDM, which was characterized by the enriched Proteobacteria and decreased Bacteroidetes.Additionally, the altered microbes, as well as the microbial metabolomics, correlated with the dosage of TAC.Specifically, the levels of beneficial microbes associated with PTDM were lowered in recipients with the high TAC trough concentrations (>5 ng·mL-1) than those with low ones (<5 ng·mL-1), which was accompanied by reduced faecal metabolites involved in the biosynthesis of α-linolenic acid and arachidonic acid-lowering factors of developing T2DM.Moreover,these microbial signatures linked with the extent of glucose disorders in LT recipients.In summary,the faecal microbiome and metabolome differed between PTDM and non-PTDM patients, which were linked with TAC dosage.This study was the first to explore taxonomic alterations and bacterial gene functions to better understand the contribution of the IM to PTDM.
1.Introduction
Post-transplant diabetes mellitus(PTDM)is a common and serious complication after solid organ transplantation (SOT), such as liver and kidney transplantation[1,2].For example,approximately 60%-90% of liver transplant (LT) recipients develop hyperglycaemia during the early post-transplant period [3,4], and 25%develop PTDM in the long term [4,5], which greatly increases the morbidity (e.g.,infection and cardiovascular events)and mortality of recipients [1].In order to avoid the misnomer of recipients of organ transplants who have undiagnosed diabetes mellitus before transplantation, the term PTDM, previously called new-onset diabetes after organ transplantation (NODAT), was adopted in 2014 to refer to time of diagnosis rather than time of occurrence [1].The acknowledged contributors to PTDM include nonmodifiable factors (such as age, race, and family history) and modifiable factors (such as the use of immunosuppressive agents) [6].The use of maintenance immunosuppressive agents, such as tacrolimus(TAC), is crucial for graft health and recipient survival [7].However, the long-term use of immunosuppressive agents is reported to be associated with risk for the development of PTDM [8,9].Our previous clinical research and other experimental studies showed that immunosuppressive agents (e.g., TAC and sirolimus),antibiotic use, and transplantation procedures caused dysbiosis of the gut microbiome [10-12], which might contribute to the development of PTDM.
In recent decades, increasing evidence has proven that the intestinal microbiota (IM) plays a crucial role in the immunologic and metabolic homeostasis of the host and the intestinal dysbiosis is involved in the development of diabetes [13,14].The bidirectional interactions between immunosuppressive therapy and the IM have been considered contributors to clinical consequences in patients [15].More recently, an experimental study showed that the TAC-induced changes in the IM were involved in the regulation of hyperglycaemia [16].Intestinal dysbiosis disrupts intestinal homeostasis and affects gut-derived metabolites, the integral immune response, and related metabolic disorders, via the ‘‘gutliver axis”, which could affect graft health in recipients[15,17,18].However,the taxonomic changes in the human IM that are related to PTDM progression remain unknown.
PTDM shares clinical characteristics with classic type 2 diabetes mellitus (T2DM), including insulin resistance, decompensated insulin release, and low-grade inflammation [1,9].Notably, our preliminary clinical findings showed that the changes in the IM in LT recipients shared similar traits with diabetes and included an increase in the levels of opportunistic pathogens and a decrease in the levels of butyrate-producing bacteria [19,20].However,intestinal dysbiosis is previously defined as unbalanced microbial components and is associated with post-transplant infections or graft function loss [21,22].Few recent experiments have shown that TAC can induce intestinal dysbiosis and be correlated with glucose disorders [12,23,24].In addition, probiotics, prebiotics, or antibiotics that modulate the IM have been shown to impact TAC-induced glucose disorders in murine experimental studies[11,16], suggesting that the IM contributes to PTDM.Thus, the alterations in microbial gene functions related to taxonomic changes in the IM need to be explored in human PTDM patients.
Here, we combined high-throughput shotgun metagenomics sequencing and metabolomics profiling targeting the whole genome and metabolites, respectively, to characterize the faecal microbial communities and metabolic molecules in LT recipients with PTDM.We further applied omics-driven bioinformatics to identify specific microbial lineages and potential functional metabolites that may contribute to the development of PTDM in recipients.This is the first metagenomics and metabolomics study of the IM in LT recipients with PTDM.Our novel findings provide an insightful understanding of the faecal microbiota in patients with PTDM and facilitate efforts to better understand the pathogenesis and microbial-targeted management of PTDM.
2.Materials and methods
2.1.Study design and enrolled patients
The subjects underwent primary liver transplantation between 2017 and 2019 in our center with at least 6 months of follow-up.All grafts were from the national donation system.The study was approved by Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University (2017-333 and 2017-425).The procedures were in accordance with regulations on human organ transplant,national legal requirements,and the Helsinki Declaration.Each recipient provided written informed consent and information such as diet, agent use, and alcohol consumption.The inclusion criteria were primary LT, stable blood concentration of immunosuppressive agents, and home-stay condition.The exclusion criteria were multiorgan transplant, less than 6 months of follow-up, and incomplete clinical data.
Faecal samples and patient information were collected during periodic outpatient follow-up appointments.Two independent cohorts were included: a cross-observational cohort of 134 recipients and a follow-up cohort of 15 recipients.Clinical data, including biochemistry indices related to glucose metabolism and TAC trough concentration (TC), were collected.The TAC TC means the valley point concentration of TAC, which is currently detected for routine monitoring for recipients.The conventional and regulatory targets for TAC TCs used in immunosuppression trials in LT were maintained at 5-15 g·L-1(i.e., 10-15 ng·mL-1during the first 4-6 weeks and 5-10 ng·mL-1thereafter) [25,26].
The LT recipients were considered to have PTDM when they had a fasting glucose level of ≥7.0 mmol·L-1, a non-fasting glucose level of ≥11.1 mmol·L-1confirmed on at least two occasions or a need for antidiabetic agents after the first post-transplant month[4].A total of 55 recipients in the discovery cohort were excluded for the following reasons: ①presence of severe complications,such as multiorgan dysfunction syndrome(MODS),sepsis, tumour recurrence, biliary stricture, and biliary and/or vascular complications through measuring serum C-reactive protein and procalcitonin levels, imaging examination, and so forth; ②infection with human immunodeficiency virus, hepatitis C virus, or other types of hepatitis virus except hepatitis B virus;③presence of organ failure or any other organ-specific diseases, including intestinal diseases and pancreatic diseases; ④ consumption of alcohol,tobacco, Chinese herbal medicine, and/or recreational drugs; and⑤re-transplantation or loss to follow-up.In the end, the study cohorts were composed of a cross-observational cohort of the 79 LT recipients, including 25 with PTDM and 54 without PTDM,and a follow-up cohort(independent)of 15 recipients treated with TAC(Fig.1 and Table 1[27]).Further,the metagenomics data from a cohort of 47 T2DM patients were included to perform the feacal microbiota difference analysis between PTDM and T2DM [28].
2.2.Sample collection, preparation, and DNA extraction
Fresh stool samples were collected,quick-frozen in liquid nitrogen,and then stored at-80°C until analysis as previously described[29].For metagenomics analysis, total faecal genomic DNA was extracted with a QIAamp®fast DNA Stool Mini Kit (Qiagen, Germany) according to the manufacturer’s protocol.
2.3.Metagenomics analysis
The DNA was extracted and sequenced on an Illumina HiSeq platform [30], and then 2×250-bp paired-end sequencing reads were generated.The DNA reads were assessed with KneadData(version 0.7.2) for quality control, and human contamination was removed by using bowtie2(version 2.2.6).The profiles of microbial composition and function were annotated by MetaPhlAn2(version 2.7.7) [31] and HUMAnN2 (version 0.11.2) [32] with the UniRef90 database.Biomarkers within the microbiome at the species level were explored using linear discriminant analysis effect size(LEfSe)[33].We calculated the Bray-Curtis distance of the above two indices with their relative abundance dataset to examine the β diversity of microbial composition and function using the vegdist function in the R (version 3.6.3) package vegan (version 2.6-2).Then, we conducted principal coordinate analysis (PCoA) using the function capscale in the same package.

Fig.1.Study design.A total of 149 LT recipients were enrolled.The discovery cohort of 79 LT recipients included 25 with PTDM and 54 without PTDM(non-PTDM).The faecal microbiota characteristics of 79 LT recipients were analyzed by whole shotgun metagenomics sequencing.In the TAC-related microbiota analysis,the 16 LT recipients were not followed, because of the administration of the immunosuppression sirolimus or both sirolimus and TAC.Further, the independent validation cohort of 15 LT recipients included 6 with high-TAC TCs (TAC-H, >5 ng·mL-1) and 9 treated with low-TAC TCs (TAC-L, <5 ng·mL-1).The faecal metabolites were analyzed by high-throughput untargeted metabolomics.
2.4.High-throughput untargeted metabolomics profiling
For metabolomics analysis, stool samples were prepared [34]and detected according to previously established methods[34,35].The samples were detected by a Dionex UltiMate 3000 RS ultraperformance liquid chromatography (UPLC) system according to previously established methods.Briefly, the gradient mobile phase consisted of water containing 0.1% formic acid (A)and methanol containing 0.1% formic acid (B) under electrospray ionization-positive (ESI+) mode and water and methanol containing 0.1% formic acid under ionization-negative (ESI-) mode.Mass spectrometric analysis was performed by Q Exactive HF-X mass spectrometry (MS) with a heated-ESI-II (HESI-II) ion source(Thermo Fisher Scientific, USA).
The acquisition mode was full MS with a scan range of 70-1050 m/z followed by data-dependent mass spectrometers (dd-MS2).Raw data were collected by XcaliburTM4.1 software and processed by Compound DiscovererTM3.1 software(Thermo Fisher Scientific).Orthogonal partial least squares-discriminant analysis (OPLS-DA)models were generated by SIMCA-P 13.0 (Umetrics AB, Sweden).The heatmap and bubble chart were generated by metaboanalyst†† https://www.metaboanalyst.ca..
2.5.Statistical analysis
Statistical analysis was performed by Statistical Package for the Social Sciences(SPSS)software(version 23.0)and GraphPad Prism software (version 7.0).Data were tested for normality of variable distributions and presented as the mean ± standard error of the mean (SEM) or median with 10-90th percentiles.The significance of differences between groups was analyzed by T test or Mann-Whitney test.As previously described [36], to assess the potential prediction effect of key microbes for PTDM, multivariable logistic regression models based on the relative abundance of the fecal microbiome were built in a stepwise manner (likelihood backward).The receiver operating characteristic (ROC) curve and thearea under the ROC curve (AUC) were calculated to evaluate the predictor performance of the final model.Results with a P value<0.05 were considered statistically significant.The network diagram was generated by Cytoscape software (version 3.8.0).The heatmap was generated by ImageGP†‡ https://www.metaboanalyst.ca.or MetaboAnalyst‡‡ https://www.metaboanalyst.ca..
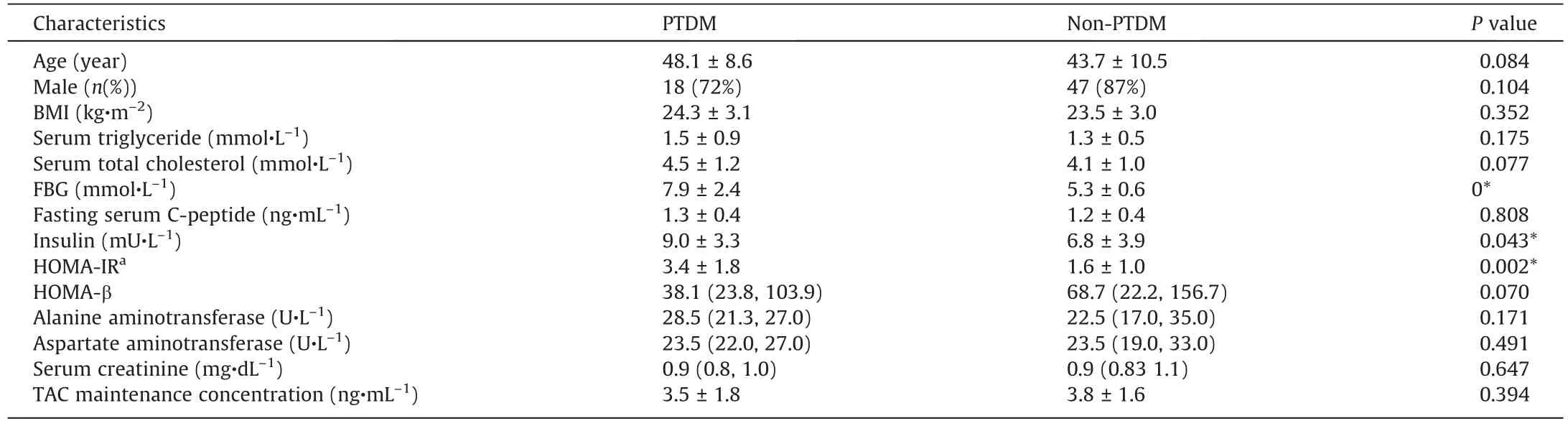
Table 1Characteristics of liver recipients with or with PTDM mellitus in discovery cohort of the study.
2.6.Data availability statement
Data availability statement raw sequence data from this experiment has been deposited in the National Center for Biotechnology Information with primary access code PRJNA687069 and SRP298863.Other data are available on reasonable request.
3.Results
3.1.Shifted IM in LT recipients with PTDM
A total of 79 LT recipients comprised the cross-observational cohort, including 25 LT recipients with PTDM and 54 non-PTDM.The detailed characteristics of LT recipients with or without PTDM were list in Table 1.Based on the shotgun metagenomics sequencing technique,we characterized the alterations of both the composition and function in the IM associated with PTDM.As shown in Fig.2(a), the distinctions in the microbial composition between these two groups were presented by the PCoA plotting.Further to identify the differentially abundant potential biomarkers for PTDM, we applied the LEfSe analysis and found that the species Paraprevotella clara, belonging to the phylum Bacteroidetes, was enriched in the non-PTDM group, while the genus Proteus, species
Proteus mirabilis and Klebsiella oxytoca, belonging to the phylum Proteobacteria, and pathogenic species Bacteroides ovatus were enriched in the PTDM group (Fig.2(b)).
Then,we performed the comparison between LT recipients with or without PTDM at the broad taxonomic levels (Fig.2(c)).Correspondingly, at the phylum level, the relative abundance of Proteobacteria was higher while the abundance of Bacteroidetes was lower in PTDM patients than that in non-PTDM group.Major differences in the phyla Proteobacteria and Bacteroidetes between PTDM and non-PTDM patients were observed from the phylum level down to lower taxonomic species levels.Interestingly, PTDM recipients showed a higher relative abundance of inflammatory class Gammaproteobacteria, order Enterobacteriales, family Enterobacteriaceae, genus Escherichia, and species Escherichia coli but a lower relative abundance of class Bacteroidia, order Bacteroidales, and species Bacteroides plebeius than those in recipients without PTDM (Fig.2(d)).
To further explore the IM feature of PTDM, we compared the shotgun metagenomics sequencing data between our cohort in this study and an independent non-transplant classical T2DM cohort from our recent study.The detail characteristics of T2DM cohort were list in Table S1 in Appendix A.There was significant segregation in the microbial composition between PTDM and T2DM groups as shown by PCoA plotting (Fig.3(a)).At the phylum level,the compositions of feacal microbiome of PTDM and T2DM groups were different (Fig.3(b)).Moreover, the relative abundance of Firmicutes and Actinobacteria was decreased while the abundance of Proteobacteria and Fusobacteria was increased in PTDM when compared with that in T2DM group (Fig.3(c)).Additionally, the LT recipient group (PTDM and non-PTDM) had the increased Proteobacteria, Bacteroidetes, Synergistetes, and decreased Firmicutes, Actinobacteria, Verrucomicrobia, when compared with T2DM group.These results demonstrated the IM of PTDM was specific and different from that of classical T2DM.
3.2.Dosage-response effect of TAC on gut microbiome in LT recipients
It is highly suggested by the mice experiments that the use of immunosuppressive TAC contributed to the glucose disorders[11,16], but remain unknown in human.Thus, to investigate the dosage-effect of TAC treatment on the IM in LT recipient cohort,we grouped the LT recipients into two groups: one with high TAC TCs (TAC-H, >5 ng·mL-1) and the other with low TAC TCs (TAC-L,<5 ng·mL-1).The characteristics of the two subgroups were list and compared (Table 2).Interestingly, we found that the higher serum insulin levels were displayed in the LT recipients of TAC-H group, supporting the hypothesis on the potential effects of TAC on glucose metabolism in LT recipients.The similar results were found in subgroups of PTDM and non-PTDM(Table S2 in Appendix A).Furthermore, we explored the potential correlation between the use of TAC and the IM dysbiosis in LT recipients.The significant differences in the β-diversity of IM between the TAC-H and TAC-L groups were indicated by the PCoA plotting (Fig.4(a)).Then, we found the distinguished microbes between the two groups by LEfSe analysis (Figs.4(b) and (c)).For instance, the taxa of the phylum Bacteroidetes, including class Bacteroidia, order Bacteroidales,family Bacteroidaceae, genus Bacteroides, species Bacteroides dorei,Bacteroides thetaiotaomicron, and other lower taxonomic levels,were enriched in TAC-H group; the taxa of the phylum Actinobacteria, including the order Bifidobacteriales, family Bifidobacteriaceae, genus Bifidobacterium, class Clostridia, genus
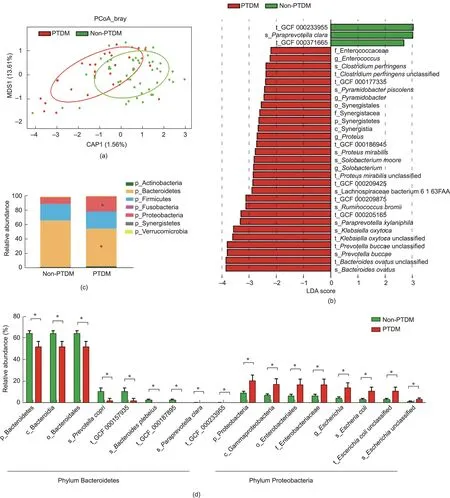
Fig.2.Differences in the faecal microbiome between LT recipients with and without PTDM.(a)PCoA score plot based on Bray-Curtis distance matrices.Each point represents one subject.The display was based on sample scores on the primary constrained axis(CAP1)and primary multidimensional scaling(MDS1).(b)LEfSe analysis showed the key discriminative biomarkers with linear discriminant analysis (LDA) score with log10 scale >2 between groups.(c) The relative abundance of faecal microbes at the phylum level.(d)The relative abundances of the phyla Bacteroidetes and Proteobacteria and their taxa.Non-PTDM,n=54;PTDM,n=25.Data are expressed as the mean±SEM.*P <0.05.p: phylum; c: class; o: order; f: family; g: genus; s: species; t: strain; GCF: assembled genomes from National Center for Biotechnology Information.
Faecalibacterium, species Faecalibacterium prausnitzii, genus Alistipes, species Alistipes putredinis, were enriched in TAC-L group.Thus, our findings suggested that TAC may induce insulin resistance in human LT recipients and further lead to PTDM via affecting gut microbial homeostasis.

Fig.3.Differences in the faecal microbiome between patients with PTDM and classical T2DM.(a) PCoA score plot based on Bray-Curtis distance matrices.Each point represents one subject.(b)The composition of faecal microbiome at the phylum level.(c)The relative abundances of the predominant phylum among PTDM,T2DM,and non-PTDM groups.Non-PTDM, n = 54; PTDM, n = 25; T2DM, n = 47.Data are expressed as the mean ± SEM.*P <0.05, **P <0.01, ***P <0.001 , ****P <0.0001.
Then, we found that the levels of most of the distinct microbes belonging to the phyla Firmicutes and Actinobacteria were lower in the TAC-H group than those in the TAC-L group, and were negatively related to the TAC TCs(Figs.4(d)and(e)).Major significant differences in the relative abundance of the phyla Firmicutes,Actinobacteria, and Bacteroidetes between the two groups were observed at broad taxonomic levels from phylum to species level(Figs.4(d)-(f)).Notably, the levels of health-beneficial bacteria,including the Bifidobacterium, Bifidobacterium longum, Bifidobacterium pseudocatenulatum, and Faecalibaterium prausnitzii, and short chain fatty acid(SCFA)-producing bacteria,including the species and genus of Alistipes,decreased in the TAC-H group and were negatively related to the TAC TCs.Moreover, the levels of most of the distinct bacteria belonging to the phylum Bacteroidetes enriched in the TAC-H group and were positively related to the TAC TCs (Fig.4(f)).Thus, the dosage-response effect of TAC on IM highly suggested a role for the use of TAC in disturbing the gut microbiome and further leading to the development of PTDM in LT recipients.
3.3.Alterations in metabolome in LT recipients with TAC treatment
To further examine the effects of TAC on the microbial gene functions related to the development of PTDM in LT recipients,we performed untargeted metabolomics analysis of feacal samples in a follow-up validation cohort of 15 LT recipients.The detailed characteristics of the validation cohort were list in Table 2.The subjects in the validation cohort were also divided into two subgroups: 9 LT recipients with low TAC TCs (TAC-L, <5 ng·mL-1),and 6 recipients with high TAC TCs (TAC-H, >5 ng·mL-1).Importantly, the faecal metabolomic profiles of the two groups wereclearly separated in the OPLS-DA plot based on the UPLC-MS data obtained from both positive and negative ion mode analysis of faecal samples (Figs.5(a) and (b)).The permutation test of the OPLSDA model supported the validation of the model(Fig.S1 in Appendix A).
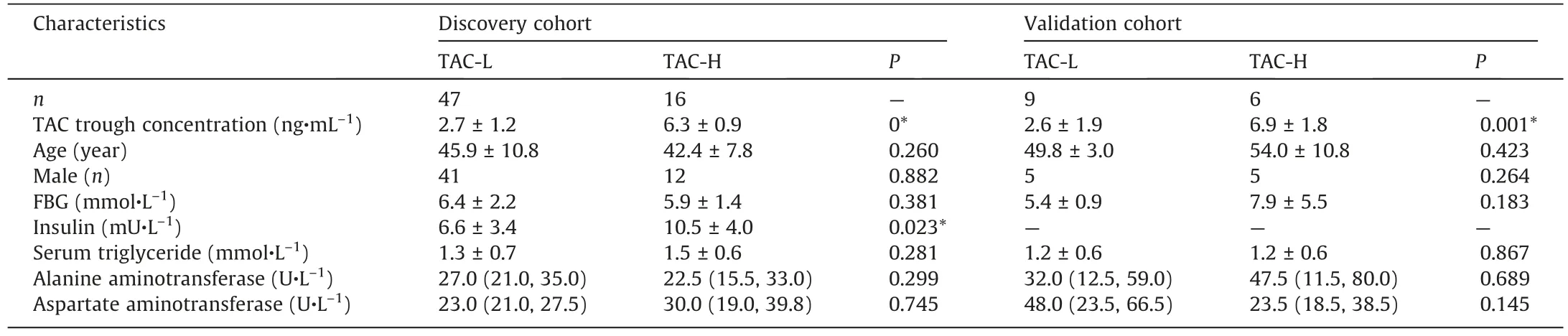
Table 2Characteristics of LT recipients grouped according to their TAC trough concentration in the study.
Further to analysis the metabolomic differences between TAC-L and TAC-H recipients, the 26 most important metabolites were identified and clustered (P <0.05) (Fig.5(c); Table S3 in Appendix A).The cluster heatmaps showed the differential metabolomic profiles between the two groups; 7 metabolites were enriched in the TAC-H group and 19 metabolites were enriched in the TAC-L group.For instance,kynurenic acid,1-linoleoyl glycerol,β-muricholic acid and phosphatidylcholine (PC) 36:4 levels were increased in the TAC-H group, while arachidonic acid, docosahexaenoic acid, (+/-)12(13)-dihydroxy-9Z-octadecenoic acid (12(13)-DiHOME), αlinolenic acid, indole-3-lactic acid, and 5-methoxysalicylic acid were enriched in the TAC-L group.
These 26 metabolites were annotated to nine metabolic pathways, mainly α-linolenic acid metabolism, arachidonic acid metabolism, biosynthesis of unsaturated fatty acids, linoleic acid metabolism, pentose and glucuronate interconversions, sphingolipid metabolism, glutathione metabolism, glycerophospholipid metabolism,and tyrosine metabolism(Fig.6(a)).More specifically,the levels of metabolites involved in α-linolenic acid metabolism and arachidonic acid metabolism were obviously lower in the TAC-H group than those of the TAC-L group, which were known to lower the risk of developing T2DM and improving insulin responsiveness; these metabolites included α-linolenic acid,(+/-)12(13)-DiHOME, (+/-)9-hydroxyoctadecadienoic acid (9-HpODE), arachidonic acid, docosapentaenoic acid, and docosahexaenoic acid (Fig.6(b)).Notably, 9 of 26 metabolites showed a significant association with the TAC TCs (Fig.6(c)).The important metabolites included arachidonic acid, 9(Z),11(E),13(E)-octadeca trienoic acid methyl ester, (+/-)12(13)-DiHOME, 16-hydroxyhexadecanoic acid, (+/-)9-HpODE, 15(R)-15-methyl prostaglandin A2, palmitoleic acid, PC 36:4, and DLdipalmitoylphosphatidylcholine.
3.4.Association between metabolic disorders and microbial dysbiosis in PTDM
To elucidate the potential role of TAC-specific microbiota in PTDM,we used the correlation network analysis and demonstrated that the discriminatory microbes were closely intercorrelated(Fig.7).Of note,the abundance of the PTDM lowered Paraprevotella clara was negatively related to the levels of Escherichia coli, Klebsiella oxytoca,and Alistipes;the levels of the TAC lowered Bifidobacterium,Bifidobacterium longum,Bifidobacterium pseudocatenulatum,Faecalibaterium prausnitzii, Alistipes, and Alistipes finegoidii were negatively related to the levels of the Escherichia coli.The results indicated that the TAC treatment might disturb some microbes and then the cascade reaction amplify the response of the whole intestinal homeostasis.These findings facilitated the role of therapeutic target on TAC-altered IM in PTDM.
To assess the association between key discriminatory microbes and glucose disorders in LT recipients, we integrated the glucose metabolism-related biochemical parameters and gut microbial profiles (Fig.8(a)).According to the heatmaps, distinct microbes of PTDM were partly associated with glucose metabolism-related biochemical parameters.In particular, the PTDM enriched microbes (such as the genus Escherichia and species Escherichia coli; species Klebsiella oxytoca, which belongs to the phylum Proteobacteria;Bacteroides ovatus,and Prevotella buccae,which belong to the phylum Bacteroidetes; species Solobacterium moorei, which belongs to the phylum Firmicutes), and the lowered ones (such as the species Paraprevotell clara, which belongs to the phylum Bacteroidetes), were positively or negatively related with the levels of HOMA-IR, FBG, and insulin, respectively.
Lastly,to assess whether the PTDM-specific microbiota could be used to classify the PTDM from non-PTDM, we performed binary logistic regression and generated predictive models.Importantly,the species Bacteroides plebeius, Paraprevotella clara, phylum Proteobacteria, and unclassified Escherichia species were selected to establish the predicted models for PTDM (Table S4 in Appendix A).The Homer-Lemeshow test indicated a good quality of data fitting (Table S5 in Appendix A).By the ROC curves analysis of the model, the faecal microbiota effectively distinguished patients with PTDM from non-PTDM (area under the curve (AUC) = 0.845,95% confidence interval (95% CI): 0.759-0.930, P <0.001) (Fig.8(b)).Notably, the predictive model based on PTDM-specific microbes and TAC-related microbes showed a better ability to differentiate patients with PTDM from non-PTDM (AUC = 0.903, 95%CI: 0.839-0.968, P = 0) (Fig.8(c); Tables S6 and S7 in Appendix A).These results highlighted the importance of the faecal microbiota in the diagnosis and treatment of PTDM.
4.Discussion
PTDM remains a risk factor for graft failure and death in SOT recipients.Here, we first reported the taxonomic and functional alterations of the human gut microbial communities in LT recipients with PTDM.More specifically, our study demonstrated the specific microbial lineages and potential functional metabolites related to TAC usage and the onset of PTDM in LT recipients.These findings strengthened the understanding of the role of intestinal dysbiosis, especially the alterations related to TAC, in PTDM and provided a novel target for the diagnosis and treatment of patients with such clinical condition.
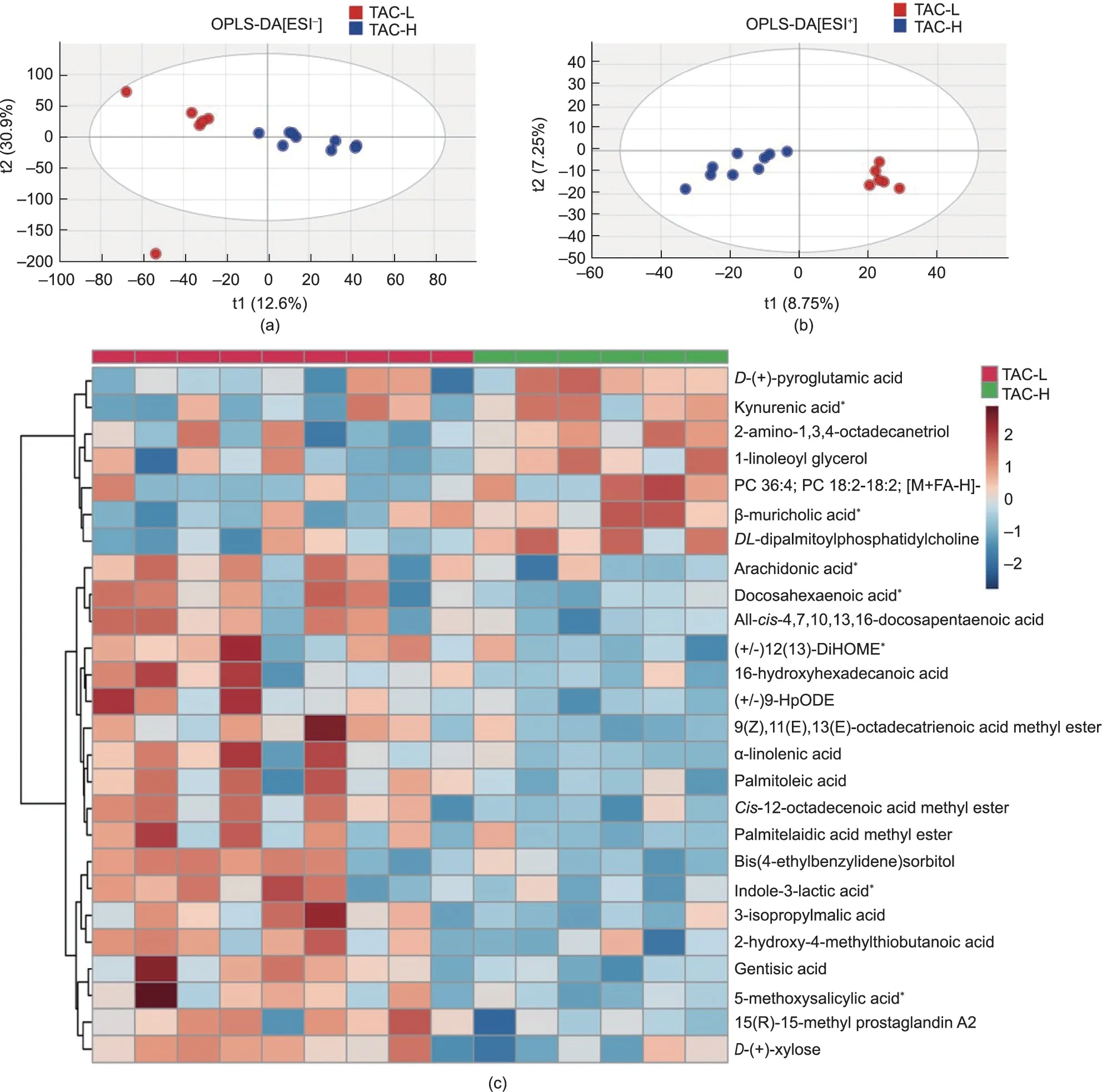
Fig.5.Microbial and nutritional metabolites in faecal metabolomics were altered by TAC usage in LT recipients.OPLS-DA plot based on the UPLC-MS/MS data in(a)positive and (b) negative ion modes.(c) Heatmap of distinct microbial metabolites between the two groups.The pentagrams represent the microbiota-related metabolites.TAC-H,n = 9; TAC-L, n = 6.MS/MS: tandem MS; PC: phosphatidylcholine; 12(13)-DiHOME: 12(13)-dihydroxy-9Z-octadecenoic acid; 9-HpODE: 9-hydroxyoctadecadienoic acid.
Here, we demonstrated that PTDM patients shared similar and special IM features with classic T2DM patients, including enrichment in the phylum Proteobacteria(such as species Escherichia coli and Proteus mirabilis) and decreased levels of the phylum Bacteroidetes (such as species Paraprevotella clara).Proteobacteria is a kind of typical Gram-negative bacteria and comprises several known human pathogens, such as Escherichia and Proteus, which have been regarded as common factors in human diseases,including metabolic disorders and inflammatory diseases[37].Therefore,Proteus mirabilis is an opportunistic pathogen and often causes infection in immunocompromised hosts[38],such as patients with diabetes or those who have immunosuppressive agents.Additionally,the level of enriched Proteobacteria was correlated with other complications (e.g., acute rejection [39] and mortality [40] after transplantation).Furthermore, our results showed that the levels of the phylum Proteobacteria and its subtaxa,especially the genus
Escherichia, species Escherichia coli, and species Klebsiella oxytoca,were positively related to glucose disorders in LT recipients.Consistently, proinflammatory Proteobacteria members flourished and were implicated in classic T2DM patients[41,42].Thus,PTDM shared similar IM features with classic T2DM, especially enrichment in pathogenic Proteobacteria.
Notably,we found that Paraprevotella clara had decreased levels while Proteus mirabilis was enriched in PTDM patients, indicating that the two species might be transplant-associated microbes.Currently,little is known about the function of Paraprevotella clara,but it is associated with a good prognosis of liver cirrhosis [43].Paraprevotella clara can utilize various sugars and produce acetic acids[44]; the latter is the smallest short-chain fatty acid (SCFA) and usually displays metabolic benefits [45,46].In our study, the decreased level of Paraprevotella clara contributed to inhibited pathways in PTDM patients,such as amino acid biosynthesis(proteinogenic amino acid biosynthesis), fermentation (pyruvate fermentation to isobutanol) and cofactor, carrier and vitamin biosynthesis(coenzyme A biosynthesis).Lower activation of amino acid biosynthesis was also reported in patients with T2DM intolerant to metformin [47].Dysregulation of coenzyme A biosynthesis presents deleterious consequences, which have been noted to occur in several pathological conditions, including diabetes and diabetic kidney disease [48,49].Moreover, our results showed a negative association between Paraprevotella clara abundance and the levels of insulin and glucose and HOMA-IR scores in LT recipients.Additionally, the PTDM patients presented higher diabetogenic function of the microbiome.The activated functional pathways included the γ-glutamyl cycle[50],enterobactin biosynthesis [51], carboxylate degradation, carbohydrate biosynthesis(sugar biosynthesis) [52,53], and fatty acid and lipid biosynthesis,which were consistent with typical T2DM.Taken together, these findings suggest that we further identified PTDM-specific microbes, including Proteus mirabilis and Paraprevotella clara.The alterations in the IM, especially the depletion of Paraprevotella clara, in addition to their functional pathways partly accounted for the disease aetiology of PTDM in LT recipients.
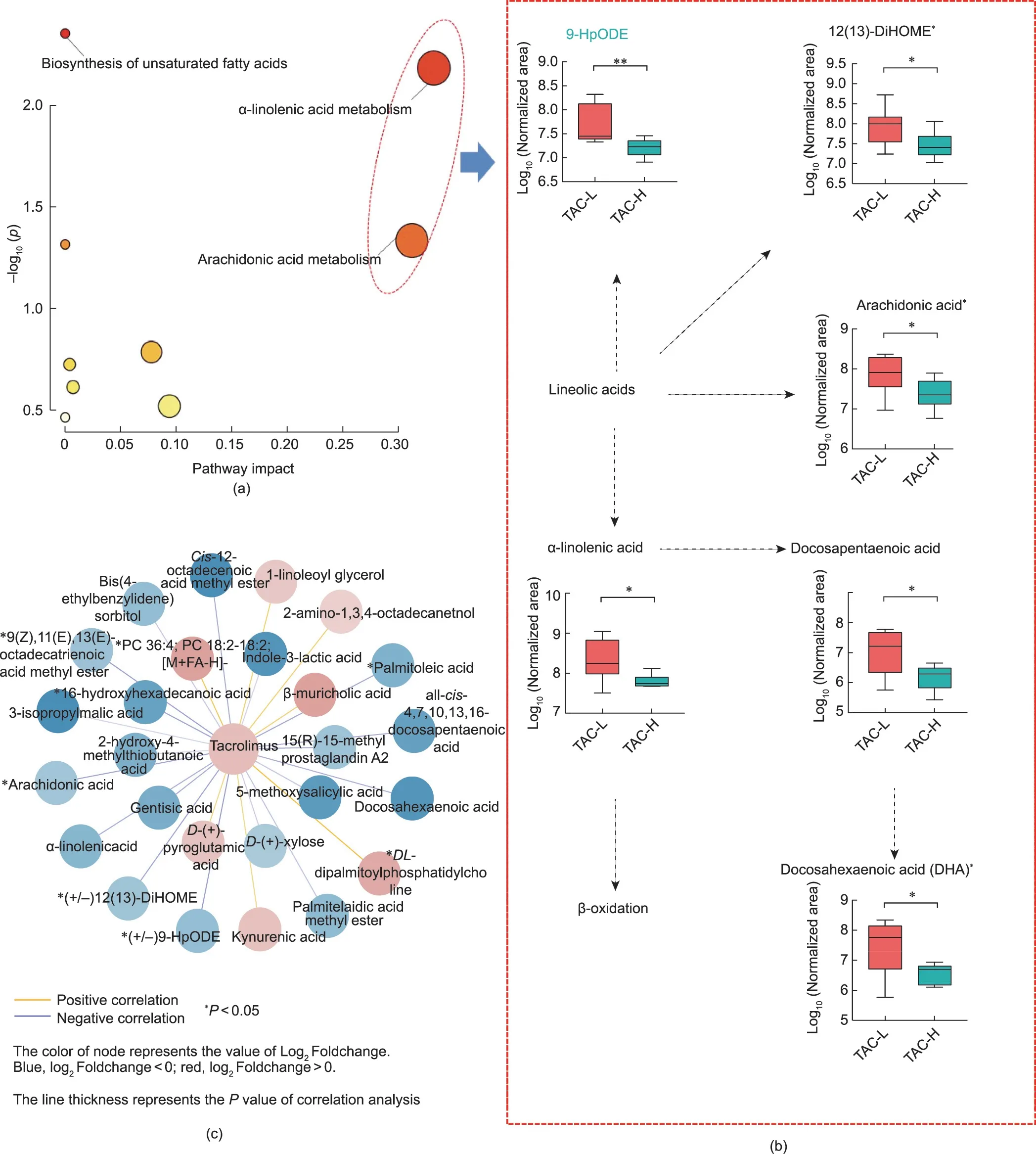
Fig.6.The signature metabolites associated with PTDM were enriched and connected with the dose of TAC in LT recipients.(a)Bubble chart based on the pathways.The sizes of the circles indicate the impact of the pathway.The color of the circles indicates the-log10p.(b)The alteration of metabolites in the pathway of α-linolenic acid metabolism and arachidonic acid metabolism.(c) Association between distinct microbial metabolites and TAC TC.*P <0.05.
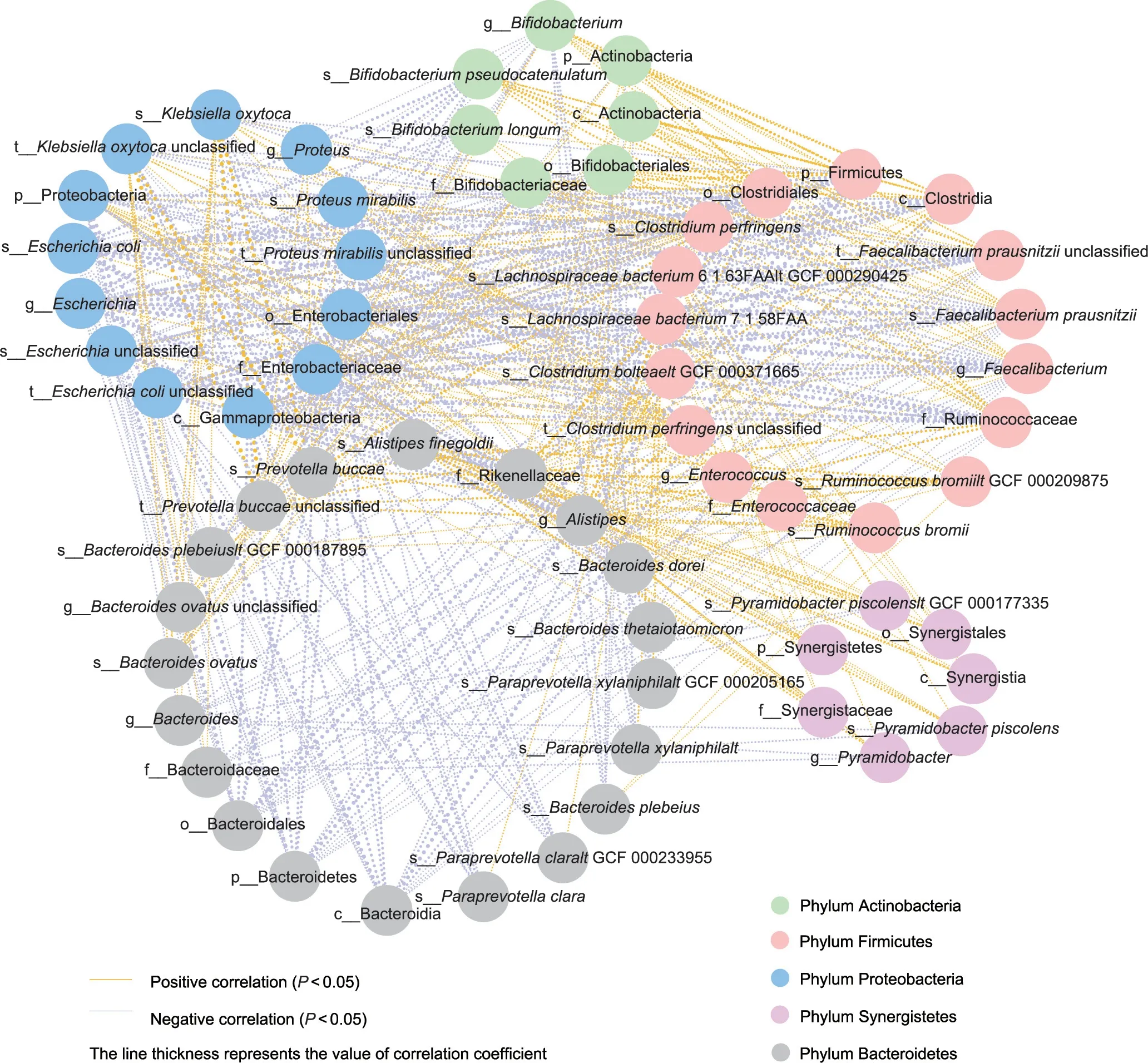
Fig.7.Distinct microbes interacted with each other in LT recipients.The line thickness represents the value of the Spearman correlation coefficient.Microbes are represented as circles, and the same color means that microbes belong to the same phylum.
To further explore the potential similarities and differences in IM between PTDM and T2DM, we made a direct comparison between PTDM patients and an independent non-transplant T2DM cohort.We found a significant impact of immunosuppressive agent in the IM of PTDM patients as compared to T2DM patients.First, PTDM showed dramatically enriched levels of the phylum Proteobacteria, which is pathogenic and could cause adverse outcome.In agreement with our findings, patients receiving SOT followed by immunosuppressive therapy, particularly mycophenolate mofetil,led to a consistent increase in Proteobacteria [15].Second, PTDM presented sharply decreased levels of Verrucomicrobia and Firmicutes but an increased level of Bacteroidetes, which was exactly in consistent with the TACinduced IM feature in our previously treated mice with TAC[11,17].Therefore, besides the common diabetic IM feature, the IM of PTDM was greatly impacted by immunosuppressive agent such as TAC as compared to that of T2DM.
Since TAC administration was implicated in the intestinal dysbiosis of liver recipients and may be associated with IM feature of PTDM, we further evaluated the correlation between IM and TAC.Interestingly, the levels of potential beneficial bacteria decreased as the TAC concentration increased.TAC, as the main immunosuppressant, was regarded as a risk factor for PTDM[54,55].Previous animal studies showed that the gut microbiome tended to be influenced by TAC [56,57] and further participated in glucose disorders [11,16,58].Our findings supported the negative association between TAC use and potential beneficial species(Bifidobacterium longum, Bifidobacterium pseudocatenulatum, Alistipes finegoldii, and Faecalibacterium prausnitzii) in liver recipients.Generally,Bifidobacterium elicits host health benefits and is widely used as a probiotic [59], of which Bifidobacterium longum and Bifidobacterium pseudocatenulatum contribute to the metabolism of dietary carbohydrates [60].Bifidobacterium abundance was reduced in mice with diabetes induced by another immunosuppressant, rapamycin [61].Moreover, the abundance of Faecalibacterium prausnitzii, as a candidate for next-generation probiotics[62], decreased in T2DM patients [41].In addition, decreased Alistipes abundance showed causally negative effects on blood triglycerides in humans [63], and contributed to fibrotic conditions in liver [64], the main organ of glucose metabolism.Our results supported that the reduction in the levels of beneficial bacteria mediated by TAC affects other commensal microbes in transplant recipients.Additionally, a panel of key microbes, including TACaltered Bifidobacteriales, could effectively discriminate patients with and without PTDM.That is, TAC altered the gut microbiome,which could be a plausible contributor to the development of PTDM-specific intestinal dysbiosis in liver recipients and further potentially influence the physiological phenotype of diabetes.
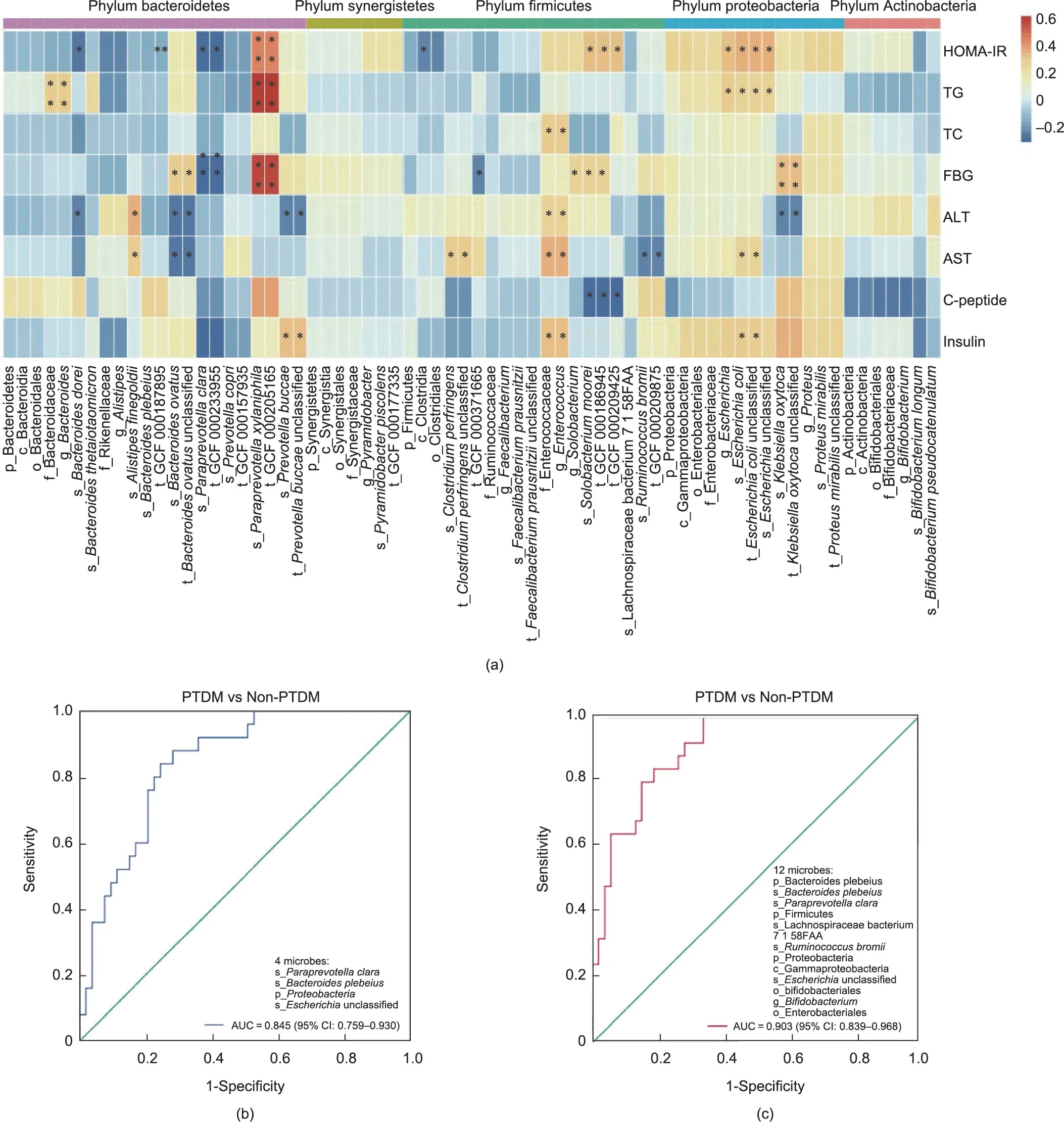
Fig.8.Distinct microbiota connected with glucose metabolic parameters in LT recipients.(a)Heatmap of correlation analysis between the microbiota and glucose metabolic parameters.(b)ROC curve of the model in discriminating PTDM from non-PTDM based on the PTDM-specific microbiota.(c)ROC curve of the model in discriminating PTDM from non-PTDM based on the PTDM-specific and TAC-altered microbiota.HOMA-IR=fasting glucose(mmol·L-1)×fasting insulin(mU·L-1)/22.5.*P <0.05.TG:triglycerides;TC: total cholesterol; ALT: alanine aminotransferase; AST: aspartate aminotransferase.
Furthermore,metabolomics analysis of the study confirmed the metabolic functional alterations of the microbiota mediated by TAC.The levels of these metabolites involved in pathways of αlinolenic acid or arachidonic acid metabolism, including arachidonic acid and 12(13)-DiHOME, were decreased in our transplant recipients with higher TAC TCs.Consistently, in T2DM patients,plasma 12(13)-DiHOME levels were negatively related to glycosylated haemoglobin levels (HbA1c, an indicator of glucose levels) and insulin sensitivity [65].12(13)-DiHOME has been identified as a promising therapeutic target for metabolic diseases,including diabetes [65,66].Additionally, it could increase skeletal muscle fatty acid oxidation, which was associated with the improvement of glucose tolerance[67].Moreover,α-linolenic acid metabolism was involved in the synthesis of unsaturated fatty acids such as docosahexaenoic acid, which could exert anti-inflammatory properties [68] and attenuate hyperglycemia via microbiota-gut-organs axis in mice [69].Arachidonic acid(metabolism) is involved in the process of inflammation [70], and the latter is an important pathogenic pathway in the development of diabetes and its complications [71].Additionally, the metabolites kynurenic acid [72], arachidonic acid [73], docosahexaenoic acid [69], (+/-)12(13)-DiHOME [74], indole-3-lactic acid [72],β-muricholic acid [75], and 5-methoxysalicylic acid could be regulated or metabolized by the microbiota.The above microbial functional alterations of inflammatory or nutrient-related metabolites associated with TAC use might partly explain the potential pathogenesis of PTDM.
This study has the limitations.First,the validation study sample was small.Our findings need to be verified in some large cohorts even with different races.Second, the patient characteristics between PTDM and T2DM had differences due to the natural history of diseases, which could be further investigated.In addition,although we revealed a PTDM-specific IM and metabolic feature,further well-designed experiment is in need to clarify the causeeffect relationships among IM, TAC, and PTDM.
In summary, this integrated metagenomics and metabolomics study firstly characterized the gut microbiome and metabolome in human LT recipients with PTDM and demonstrated that the altered gut microbial features in LT recipients were closely correlated with the serum concentration of TAC.Additionally,the prevalence of certain Proteobacteria, and the decreased abundance of beneficial microbes (e.g., Bifidobacterium and Faecalibacterium), in addition to disturbed energy metabolism in LT recipients may affect prognosis.As we previously showed a negative impact of antibiotics on TAC-induced glucose disorders[11],we hypothesize a beneficial role of prebiotics and probiotics in the treatment of PTDM.Although it is difficult to unequivocally draw the causal association between specific microbes and PTDM occurrence in LT recipients based on current results, the findings of the study supported a role of the gut microbiota in PTDM patients, which could potentially serve as the signature and adjuvant preventive and therapeutic target for glucose disorders in clinical recipients.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82170668, 82171757, and 82241215), the National Key Research and Development Program of China(2021YFA1301001), the Sino-German Center for Research Promotion (GZ1546), the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2019-I2M-5-045), and the Research Project of Jinan Microecological Biomedicine Shandong Laboratory (JNL-2022040C and JNL-2023006C).
Authors’ Contribution
Baohong Wang designed and managed the experiments;Baohong Wang, Qi Ling, and Yuqiu Han wrote the manuscript; Qi Ling, Yuqiu Han, Xiaosen Wang, Zheng Zhu, Jingyu Wang, Jiaying Cao, and Xiaohan Lin performed the experiments; Yuqiu Han,Yue Ma,Baohong Wang,and Jun Wang analyzed the data;Baohong Wang, Qi Ling, and Jun Wang revised the manuscript.
Compliance with ethics guidelines
Qi Ling, Yuqiu Han, Yue Ma, Xiaosen Wang, Zheng Zhu, Jingyu Wang, Jiaying Cao, Lin Xiaohan, Jun Wang, and Baohong Wang declare that they have no conflict of interest or financial conflicts to disclose.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2023.09.006.
杂志排行

Engineering的其它文章
- Global Top Ten Engineering Achivements 2023
- 2023 Global Engineering Fronts
- Will Massive Appetite for Minerals Stall Clean Energy Transition?
- Optical Microscopy Advances Reach Sub-Nanometer Resolution
- International Correlation Research Program: Cross-Fault Measurement for Earthquake Prediction
- A Systematic Perspective on Communication Innovations Toward 6G