Profiling the Antimalarial Mechanism of Artemisinin by Identifying Crucial Target Proteins
2023-03-22PengGoJinyouWngJiyunChenLiweiGuChenWngLitingXuYinKwnWongHuiminZhngChenghoXuLingyunDiJigngWng
Peng Go, Jinyou Wng, Jiyun Chen, Liwei Gu, Chen Wng, Liting Xu, Yin Kwn Wong,Huimin Zhng, Chengho Xu,*, Lingyun Di*, Jigng Wng,b,,*
a State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Artemisinin Research Center, and Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China
b Pharmaceutical College, Henan University, Kaifeng 475004, China
c Shandong Academy of Chinese Medicine, Jinan 250014, China
d Department of Infectious Disease, Shenzhen Clinical Research Centre for Geriatrics, Shenzhen People’s Hospital & the First Affiliated Hospital of Southern University of Science and Technology, Shenzhen 518020, China
Keywords:Artemisinin Antimalaria Target identification MS-CETSA Transcriptomics
ABSTRACT The widespread use of artemisinin(ART)and its derivatives has significantly reduced the global burden of malaria; however, malaria still poses a serious threat to global health.Although significant progress has been achieved in elucidating the antimalarial mechanisms of ART, the most crucial target proteins and pathways of ART remain unknown.Knowledge on the exact antimalarial mechanisms of ART is urgently needed, as signs of emerging ART resistance have been observed in some regions of the world.Here, we used a combined strategy involving mass spectrometry-coupled cellular thermal shift assay (MS-CETSA)and transcriptomics profiling to identify a group of putative antimalarial targets of ART.We then conducted a series of validation experiments on five prospective protein targets, demonstrating that ART may function against malaria parasites by interfering with redox homeostasis, lipid metabolism, and protein synthesis processes.Taken together, this study provides fresh perspectives on the antimalarial mechanisms of ART and identifies several crucial proteins involved in parasite survival that can be targeted to combat malaria.
1.Introduction
Malaria remains one of the most lethal infectious diseases worldwide and was responsible for approximately 247 million infection cases and 619 000 deaths in 2021 [1,2].The use of artemisinin(ART)and its derivatives as well as ART-based combination therapies (ACTs) has markedly reduced the global malaria burden[3].ART is the only first-line antimalarial drug that does not involve widespread drug resistance at present [2].However, the emergence of ART and ACT resistance in some regions of the world has triggered concerns over the continued effectiveness of ART in malaria control [4,5].
After many decades, researchers in the field have started to unravel the mechanisms of action behind ART’s antimalarial effect[6], especially with the applications of various-omics technologies in recent years[7-9].Our group was among the first few laboratories in the world to use the activity-based protein profiling(ABPP)technique to report the promiscuous targeting of ART [10].The unique antimalarial effect of ART originates from the hyperactivation of its peroxide bridge by heme, an unavoidable byproduct from the hemoglobin metabolism of parasites in erythrocytes.Subsequently, massive amounts of free radicals are produced and can alkylate a large number of plasmodium proteins and biomolecules,leading to rapid parasite death [11].In addition to our work, evidence from several other studies has also supported the seemingly nonspecific mode of action of ART [12,13].Instead, the specificity of ART appears to be mediated by the specific parasite growth stage, i.e., the asexual intraerythrocytic stage when the parasites replicate in erythrocytes and require hemoglobin.Having identified this unique mode of action, we were next interested in determining the specific biological processes and target proteins of ART that play the most crucial roles in its antimalarial activity and parasite survival.The identification of these pathways and protein targets will not only strengthen our knowledge of ART usage in malaria control but will also shed light on novel and specific directions for tackling ART resistance.
In this work,the isothermal dose-response format(ITDR)of the mass spectrometry (MS)-coupled cellular thermal shift assay(CETSA), known as ITDR-MS-CETSA, was first used to identify potential target proteins of artesunate (ATS), a commonly used ART derivative with much better aqueous solubility and similarly high parasiticidal ability as ART[10].CETSA is a label-free drug target deconvolution technology in which the original unmodified drug molecules are used [14,15].We and others have previously used the ITDR-MS-CETSA method to study the target proteins and working mechanisms of antimalaria drugs, including quinine and chloroquine[16,17].We also used the transcriptomics method to profile the modulated gene expression after ATS treatment.By analyzing the obtained data,we then carried out a series of validation experiments on several potential hit proteins involved in the oxidative stress response, lipid metabolism, and protein synthesis of Plasmodium falciparum (P.falciparum) parasites, demonstrating that these pathways could play important roles in parasite survival and underlie the antimalarial function of ART.
2.Materials and methods
2.1.Reagents
AlbumiNZTMbovine albumin low endotoxin (#0219989680)was purchased from MP Biomedicals(USA).Roswell Park Memorial Institute (RPMI) 1640 (#31800022) was purchased from Gibco(New Zealand).Other reagents used for malaria parasite culture were purchased from Sigma-Aldrich (USA).
Reagents used for CETSA and other biochemical experiments were as follows: 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfo nic acid (#0511) was purchased from Avantor (USA).β-glycerophosphate (#G9422), Na3VO4(#S6508), MgCl2(#M2670), tris(2-carboxyethyl)phosphine (TCEP; #SLCB2088),ammonium formate (#70221), chloroacetamide (CAA; #22790),and trifluoroacetic acid (#T6508) were purchased from Sigma-Aldrich.Tris(hydroxymethyl)aminomethane(#31801),ethylenediaminetetraacetic acid-free protease inhibitor cocktail (#1861278),Pierce BCA Protein Assay Kit (#23228), triethylammonium bicarbonate buffer (TEAB; #90114), 0.1% formic acid in water(#192741), acetonitrile (#51101), and TMT10plex isobaric label reagent (#90110) were purchased from Thermo Fisher Scientific(USA).Lysyl endopeptidase (LysC; #125-05061) was purchased from Fujifilm(Japan); RapiGest SF (#191891) was purchased from Waters (USA).Trypsin (#V5280) was purchased from Promega(USA).All water used in the study was obtained from an ELGA PURELAB Classic ultra-pure water system (UK).
2.2.Parasite culture
The P.falciparum (3D7 strain) was cultured based on the method established previously with slight modifications [17,18].Parasites were cultured in malaria complete medium supplemented with 2% healthy human erythrocytes and placed in a constant temperature three-gas incubator at 37°C,5%O2,5%CO2,and 90% N2dynamically on a circular shaker.Parasite cultures were synchronized twice by treatment with 5% sorbitol at 37 °C for 10 min.The parasitemia was assessed daily using Giemsa-stained thin blood smears and maintained at 5%-10%.
2.3.Target identification by ITDR-MS-CETSA
The experiment was performed similarly as in our prior study with minor modifications[17].Unsynchronized P.falciparum were continuously cultured to 10% parasitemia at 2% hematocrit and then harvested.Different concentrations of ATS (0-100 μmol·L-1)were incubated with equal volumes of extracted parasite lysate.Next, the samples were divided into two equal portions, heated at 37 or 52 °C for 3 min, and then cooled at 4 °C for 3 min.The soluble proteins were collected after centrifugation at 14 000 r·min-1for 20 min.Soluble proteins for each sample were reduced with 20 mmol·L-1TCEP and 0.05% RapiGest in 100 mmol·L-1TEAB for 20 min at 55 °C and then alkylated with 55 mmol·L-12-CAA for 30 min in the dark at room temperature(RT).After digestion with LysC and trypsin,the samples were centrifuged at 14 000 r·min-1for 20 min, and the supernatant was dried using a centrifugal vacuum evaporator.The peptide samples were labeled with TMT10plex reagents for 2 h at RT and then desalted using an Oasis hydrophilic-lipophilic-balanced (HLB)column.Peptide offline prefractionation was performed using a Nexera LC-40D XS liquid chromatography(LC)system(SHIMADZU,Japan).All fractions were collected and pooled into 20 fractions for LC-tandem MS (LC-MS/MS) analysis.
2.4.LC-MS/MS measurement and protein identification
LC-MS/MS analyses were performed as described previously[17].Dried peptide sample fractions were reconstituted and separated on a C18 high-performance LC reversed-phase analytical column.LC-MS/MS data acquisition was conducted in data-dependent acquisition mode.The acquired spectra were analyzed using Proteome Discoverer (PD, version 2.4) against the P.falciparum 3D7 protein database (PlasmoDB-58)†‡ https://github.com/nkdailingyun/mineCETSA..The specific search parameters used in PD version 2.4 were the same as those described in our prior study [17].
2.5.ITDR data processing and bioinformatics analysis
The data analysis was carried out with R statistical software(version 4.2.0) with the mineCETSA package‡‡ https://github.com/nkdailingyun/mineCETSA.similarly as previously described [17].Gene Ontology (GO) analysis was conducted with the clusterProfiler package of R statistical software with a significance threshold at a false discovery rate (FDR) <0.05.
2.6.Sample preparation for transcriptome and metabolomics profiling
The status and infection rate of parasites were assessed daily using Giemsa staining.Unsynchronized parasites were cultured to 10% parasitemia and then treated with ATS (600 nmol·L-1) and equal volumes of dimethyl sulfoxide (DMSO) vehicle (0.1% final concentration) for 5 h in continuous culture.Afterward, infected erythrocytes were immediately collected and lysed with ten-fold volumes of 0.05% saponin on ice to release parasites.
And then this morning, I walked you up the steep hill to your classroom with a picture of the president on one wall and of Bambi on the opposite. You found the coat hook with your name above it right away, and you gave me one of your characteristically fierce, too-tight hugs. This time you were ready to let go before I was.
2.7.RNA-sequencing (RNA-seq) analysis and data processing
Subsequently, parasite RNA isolation, library construction, and sequencing were performed as described previously [19].The NEBNext Ultra RNA Library Prep Kit for Illumina (USA) was used to construct the sequencing library according to the manufacturer’s recommendations.Briefly, the total RNA of parasites was extracted with TRIzol RNA isolation reagent (Invitrogen, USA),and the parasite messenger RNA(mRNA)was purified using poly-T oligo-attached magnetic beads.The library fragments were purified to preferentially select complementary DNA fragments of 370-420 base pairs (bp) in length with the AMPure XP system(USA).Then,polymerase chain reaction(PCR)was performed with Index(X)Primer,Universal PCR primers,and Phusion High-Fidelity DNA polymerase.The qualified libraries were pooled and sequenced on Illumina platforms (Novogene, China).Three independent biological replicates were conducted for each treatment group.The differentially expressed genes (DEGs) were identified through the DESeq2 (version 1.10.0) R package for comparison before and after malaria parasites were treated with ART.Genes with P <0.05 and log2(FC) >0.5(FC: fold change)were considered DEGs.The bioinformatics analysis of DEGs was performed as described in Section 2.5.
2.8.Lipidomic analysis and data processing
The metabolite extraction of parasites(2×108cells per sample)was performed as previously described[20].Samples were injected into a Hypesil Gold column using a 12-min linear gradient at a 0.2 mL·min-1flow rate.The eluents were 5 mmol·L-1ammonium acetate (pH 9.0) (A) and methanol (B) for the negative polarity mode and 0.1% formic acid (A) and methanol (B) for the positive polarity mode.All experiments were repeated in duplicate and three times independently.The raw data were processed using Compound Discoverer 3.1 (Thermo Fisher Scientific).The main search parameters were as follows: retention time tolerance,0.2 min; actual mass tolerance, 5 parts per million (ppm); signal/noise ratio,3;and signal intensity tolerance,30%.Metabolites with variable importance in projection (VIP) >1, log2(FC) >1.200 or log2(FC) < 0.833, and P < 0.05 were considered differential metabolites.
2.9.Expression and purification of recombinant P.falciparum proteins
The coding sequences of P.falciparum protein genes were obtained from PlasmoDB-58.The DNA sequences were synthesized and cloned into the BamH I and Xho I sites of the pET-28a (+)expression vector by GENEWIZ (China).Plasmids were transformed into Escherichia coli BL21(DE3), and proteins were induced by 0.4 mmol·L-1isopropyl-β-D-thiogalactoside at 17 °C in Luria-Bertani medium.Cells were pelleted and resuspended in lysis buffer (20 mmol·L-1tris-HCl, pH 7.4, 200 mmol·L-1NaCl, 1 mmol·L-1phenylmethanesulfonyl fluoride) and lysed by a JN-Mini Pro cell disruptor(JNBIO,China)under 1200 bar(1 bar=100 kPa)pressure.The clarified lysate supernatant was collected and loaded onto a Ni-nitrilotriacetic acid (NTA) chromatography column.Recombinant proteins were eluted at 4 °C.
2.10.Fluorescence labeling of recombinant proteins
The validation of ART targeting to recombinant target proteins in vitro was performed as previously described[17].In brief,equal amounts of recombinant protein (2 μg) were incubated with increasing concentrations of the ART-based activity probe AP1 or with the same concentrations of AP1 for different times.For the competition assays, recombinant proteins were pretreated with excess ATS or iodoacetamide (IAA) for 2 h before being incubated with AP1 or IAA-alkynyl probe(IAA-P)for another 2 h.Click chemistry reaction-based fluorescence labeling was conducted for 2 h at RT after tetrame-thyl-6-carboxyrhodamine azide fluorescent tag(TAMRA-azide; 50 μmol·L-1), TCEP (1 mmol·L-1), tris-hydroxypropyltriazolylmethylamine (THPTA; 1 mmol·L-1), and CuSO4(1 mmol·L-1) were sequentially added.Subsequently, proteins were precipitated by prechilled acetone, dissolved in 1× sodium dodecyl sulfate (SDS)-loading buffer and separated by SDSpolyacrylamide gel electrophoresis (PAGE) electrophoresis.Fluorescence scanning was performed using a Sapphire Imager(Azure,USA), and Coomassie brilliant blue staining was also performed as a loading control.
2.11.Target validation by pull-down Western blotting
Parasites were maintained in continuous culture and then treated with AP1(with or without pretreatment with excess ATS).The soluble parasite lysates were extracted, and the click chemistry reaction was performed using CuSO4(1 mmol·L-1), TCEP(1 mmol·L-1), THPTA (100 μmol·L-1), and biotin-azide(50 μmol·L-1) to conjugate the biotin tag for 2 h at RT.Subsequently, biotinylated proteins were enriched with NeutrAvidin beads (Thermo Fisher Scientific) for 4 h.The enriched proteins were separated by SDS-PAGE, transferred onto polyvinylidene fluoride membranes,and visualized using electrochemiluminescence(ECL) after incubation with corresponding specific antibodies.
2.12.Intracellular imaging assay
The imaging assay was conducted as described in our prior research [17].The parasites were cultured in a 24-well plate with 2% hematocrit and 5% parasitemia.After treatment with AP1 or DMSO for 30 min, the parasites were fixed.Then, parasites were dripped onto coverslips precoated with 0.01% (w/w) polylysine and permeabilized.Subsequently, the click chemistry reaction was conducted with TAMRA-azide.The coverslips were then transferred onto microscope slides and imaged.For the subcellular colocalization assay, parasites were incubated with the corresponding primary and secondary antibodies and imaged after the click chemistry reaction.All colocalization experiments were performed with a Dragonfly 200 confocal microscope, and semiquantitative analysis was performed with JACoP (plugin of ImageJ).
2.13.Activity assay for recombinant P.falciparum 1-Cys peroxiredoxin(rPf1-CysPxn)
A Hydrogen Peroxide Assay Kit (Beyotime, China) was used to measure the effect of ATS on the enzymatic activities of rPf1-CysPxn proteins.First, different concentrations of ATS (0-5 μmol·L-1) were incubated with purified rPf1-CysPxn (10 μg) at 28 °C for 1 h.Subsequently, 10 μL H2O2(final 50 μmol·L-1) was added to the mixture,and the reaction was stored at RT for 30 min before an activity detection working solution was added.The absorbance of the samples at 450 nm was measured with a microplate reader(PerkinElmer, USA).Reaction buffer with ATS but no rPf1-CysPxn was used as the negative control.The experiments were carried out in triplicate.
2.14.De novo protein synthesis inhibition assay by azidohomoalanine(AHA) labeling
2.15.Identification of the binding sites of ATS on recombinant proteins
Recombinant proteins (10 μmol·L-1) were first incubated with 500 μmol·L-1ATS, 20 μmol·L-1hemin, and 200 μmol·L-1NaVc for 4 h at RT.Excess reagents were removed by Protein Desalting Spin Columns (#89849) according to the manufacturer’s instructions.Next, ammonium bicarbonate was added to a final concentration of 25 mmol·L-1, and the samples were reduced and alkylated with 10 mmol·L-1dithiothreitol(DTT)and 50 mmol·L-1IAA followed by digestion at 37 °C overnight.Samples were desalted and dried before resuspension for LC-MS/MS analysis as described previously.The acquired spectra were analyzed using pFind (version 3).The specific search parameters were set as follows: precursor and fragment tolerance were set to 20 ppm.Peptide spectra matches were filtered with false-discovery rates of 1%on the peptide spectrum match.
2.16.Molecular docking model
Molecular Operating Environment (version 2019.0102) was used for docking simulation.The chemical structure of ATS was downloaded from PubChem (ID: 5320351).The protein structure of Pf1-CysPxn was downloaded from the Protein Data Bank database and prepared using the QuickPrep module.The docking parameters were set as follows: Triangle Matcher, Rigid Receptor,initial scoring method London dG retaining 30 poses and the final scoring method used generalized-born volume integral/weighted surface area (GBVI/WSA) dG with 5 poses.The amino acid residue region within 4.5 Å was chosen as the active pocket.
3.Results and discussion
3.1.Identifying the targets of ATS using ITDR-MS-CETSA
We first applied ITDR-MS-CETSA technology to identify the direct-binding target proteins of ATS using the principle of drugbinding-induced thermal shifts (Fig.1(a)).P.falciparum lysates were prepared from the intraerythrocytic stage and then incubated with a gradient dose of ATS (0-100 μmol·L-1).The samples were challenged by a 3 min pulse of 52°C heating,and similarly treated samples at 37 °C were used as a reference control.We identified 145 potential hits out of 2749 measured proteins (Table S1 in Appendix A),with 31 and 114 proteins showing positive and negative thermal shifts,respectively(Fig.1(b)).GO enrichment and protein-protein interaction analysis of these potential target proteins suggested that they are mainly involved in the organonitrogen metabolic process,proteolysis,ribosome,translation,and catabolic process, among others (Fig.1(c); Fig.S1 in Appendix A).Notably,compared to the 124 target proteins of ART generated in our earlier study using ABPP technology, the overlap was 7.6% (Fig.S2 and Table S2 in Appendix A).There is discrepancy in the identified target proteins due to the different working principles and mass spectrometer used, which is not surprising.Nevertheless, both of these studies clearly indicated that the excellent antimalarial effect of ART may mainly stem from a collective effect produced by the interaction of ART with a number of proteins.
3.2.Transcriptomic analysis of parasites in the intraerythrocytic stage after treatment with ATS
Next, we analyzed the changes in the whole transcriptome of P.falciparum after ATS treatment by transcriptomic analysis(RNA-seq).The results showed a significant modulation of gene expression,in which 2016 genes were upregulated and 1193 genes were downregulated after drug treatment (P < 0.05 and log2(FC) >0.5) (Figs.2(a) and (b)).The GO analysis suggested that proteolysis, lipid metabolic process, and cellular protein catabolism were downregulated and that RNA metabolic process and organonitrogen compound metabolic process were upregulated(Figs.2(c) and (d)).
We then examined the expression changes in the 145 target proteins at the transcriptome level and found that 65 targets were upregulated and 57 were downregulated(Figs.3(a)and(b)).These 57 downregulated target genes are mainly involved in the protein catabolic process, organonitrogen catabolic process, and lipid biosynthetic process, while the 65 upregulated target genes are largely involved in the protein folding and response to oxidative stress processes (Fig.3(c)).
From the enriched functions/processes highlighted by our transcriptomic data, we identified three major physiological functions of interest that are potentially targeted by ATS, namely, redox homeostasis, lipid metabolism, and protein synthesis.To validate the effect of ATS on these processes, we next carried out an array of verification experiments on five representative proteins involved (marked with a red asterisk in Fig.3(d)).These proteins are known to play important roles in the functions of interest and could thus may be crucial targets of ATS in controlling malaria parasites.
3.3.ART affects the redox homeostasis of P.falciparum
At the intraerythrocytic stage,P.falciparum parasite is subjected to constant oxidative challenges due to its catabolism of host hemoglobin and the attack from the host immune system[21,22].Without real catalase and glutathione peroxidase, parasites mainly use superoxide dismutases and peroxiredoxins to maintain intercellular redox homeostasis [23].Peroxiredoxins are a family of cysteine-dependent peroxidases that can catalyze the reduction of hydrogen peroxide and organic hydroperoxides and play an essential role in enhancing parasite survival under oxidative stresses[23].Based on the number of active cysteines involved in catalysis, peroxiredoxins can be grouped into 1-Cys and 2-Cys peroxiredoxins [24].There are two 2-Cys peroxiredoxins and one 1-Cys peroxiredoxin in P.falciparum [25].
Here, we identified P.falciparum thioredoxin peroxidase-1(PfTrx-Px1, PF3D7_1438900) and Pf1-CysPxn (PF3D7_0802200) as potential targets.PfTrx-Px1 is a typical 2-Cys Peroxiredoxins protein in the cytoplasm of P.falciparum that plays an important role in detoxifying reactive oxygen species (ROS) in parasites [26,27].Notably, PfTrx-Px1 was also identified as a target protein in our earlier ABPP-based target identification study [10], although it was not further validated at that time (Table S2).Pf1-CysPxn is a cytosolic protein expressed at a high level during the intraerythrocytic stage and has been reported to be involved in heme detoxification [28].We decided to first use an ART-based chemical probe AP1 to validate the targeting of ATS to recombinant PfTrx-Px1(rPfTrx-Px1) in vitro using the ABPP principle [7,29].AP1, which was previously developed by our group, largely retains the same level of antimalaria efficacy as ATS and is modified with a small alkyne tag, permitting subsequent click chemistry reaction with an affinity or fluorescent moiety [10].The fluorescence labeling experiments suggested that AP1 could bind to the rPfTrx-Px1 proteins in a dose- and time-dependent manner (Figs.4(a) and (b)),and excessive ATS effectively competed with the binding (Fig.4(c)).Similarly, the targeting of ATS to rPf1-CysPxn was also verified (Figs.4(d)-(f)).When the two proteins were subjected to fluorescence labeling under the same conditions, the fluorescence intensity of rPf1-CysPxn was much stronger than that of rPfTrx-Px1 (Fig.4(g)), suggesting that ART preferentially targets Pf1-CysPxn over PfTrx-Px1.Furthermore, labeling could also be performed in living cells,as indicated by both immunofluorescence staining and pull-down Western blotting experiments (Figs.4(h)-(j)).In addition,the enzyme activity assay indicated that ATS could inhibit the catalytic activity of rPf1-CysPxn in a dose-dependent manner (Fig.4(k)).
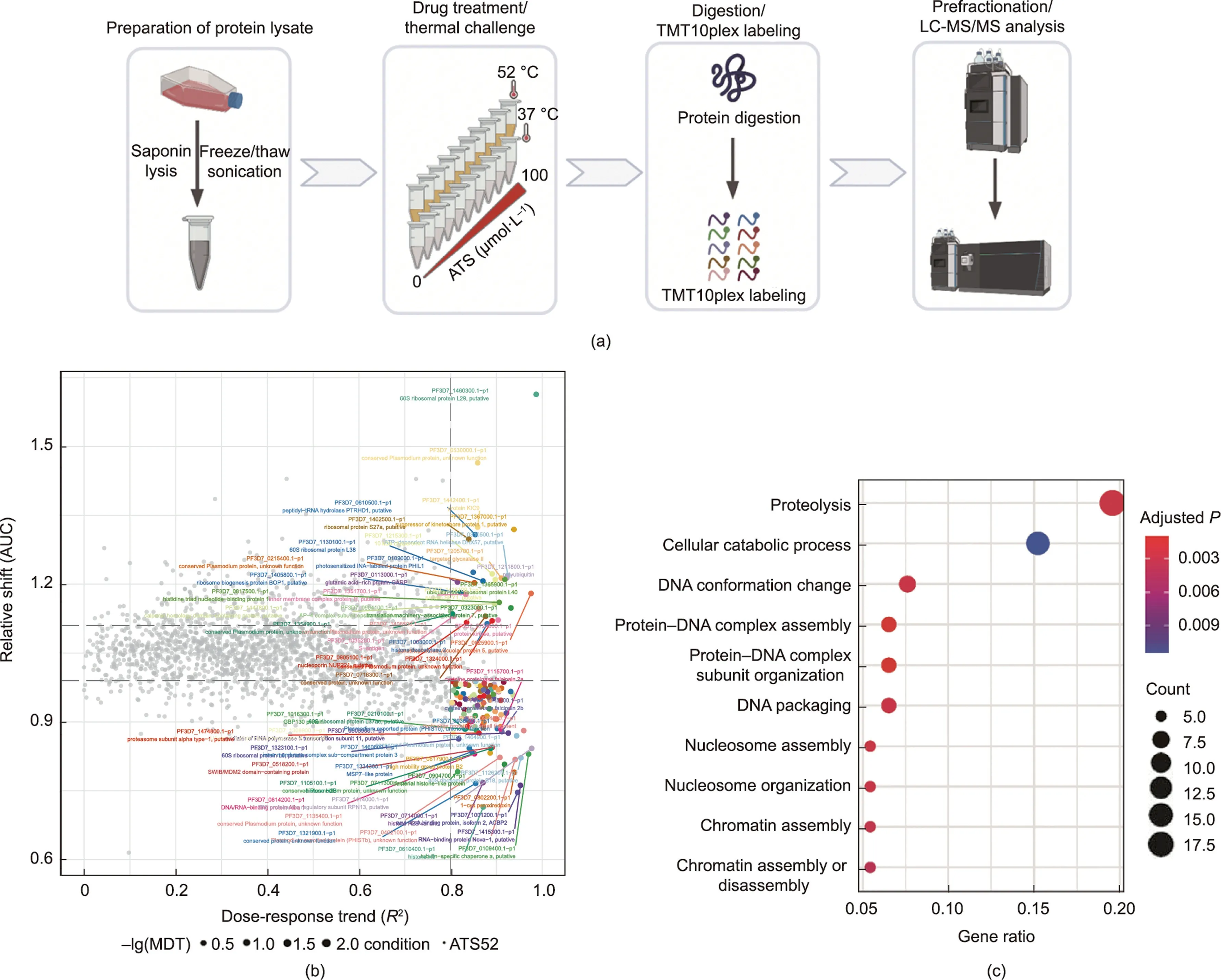
Fig.1.Identification of ATS targets with ITDR-MS-CETSA.(a)Schematic workflow of ITDR-MS-CETSA used in this work.Parasite lysates were treated with a range of different concentrations (0-100 μmol·L-1) of ATS and then heated under different temperatures.The soluble proteins were collected for subsequent analysis as described in our previous study[17].(b)The dose-response curve fitting quality(R2)-area under the curve(AUC)plot displaying the thermal stability shift in the P.falciparum proteome after treatment with ATS.The potential hits are displayed in color and annotated.(c) GO analysis of the potential hits of ATS from ITDR-MS-CETSA analysis.
We were then interested in determining whether ATS targets a specific region/site to inhibit the activities of Pf1-CysPxn.By using a cysteine-targeting IAA-P as a labeling reagent,we first confirmed that ATS can efficiently compete with the binding of IAA-P to rPfTrx-Px1 (Fig.4(l)) and rPf1-CysPxn (Fig.4(m)), suggesting that cysteines are the key targeting residues for ATS.Next, we incubated ATS with rPfTrx-Px1 and used MS to locate the binding site of ATS.The results suggested that ATS interacts with the peroxidatic cysteine 47 (C47) and phenylalanine 9 (F9) sites on rPf1-CysPxn at (Figs.4(n) and (o)), and the latter may be attributed to promiscuous binding by free alkylating radicals[30,31].Both binding sites were further validated by docking simulation (Fig.S3 in Appendix A).The labeling of AP1 was obviously diminished on the double-site mutants of rPf1-CysPxn (Fig.4(p)).Furthermore,the double-site mutant of rPf1-CysPxn showed clearly diminished enzymatic activities compared to that of the single-site mutant(F9A or C47A) and wild-type proteins (Fig.4(q)).Therefore, ATS can bind to rPf1-CysPxn and inhibit its peroxidase activity, hampering the clearance of damaging ROS.
3.4.ART interferes with the biogenesis of phosphatidylcholine
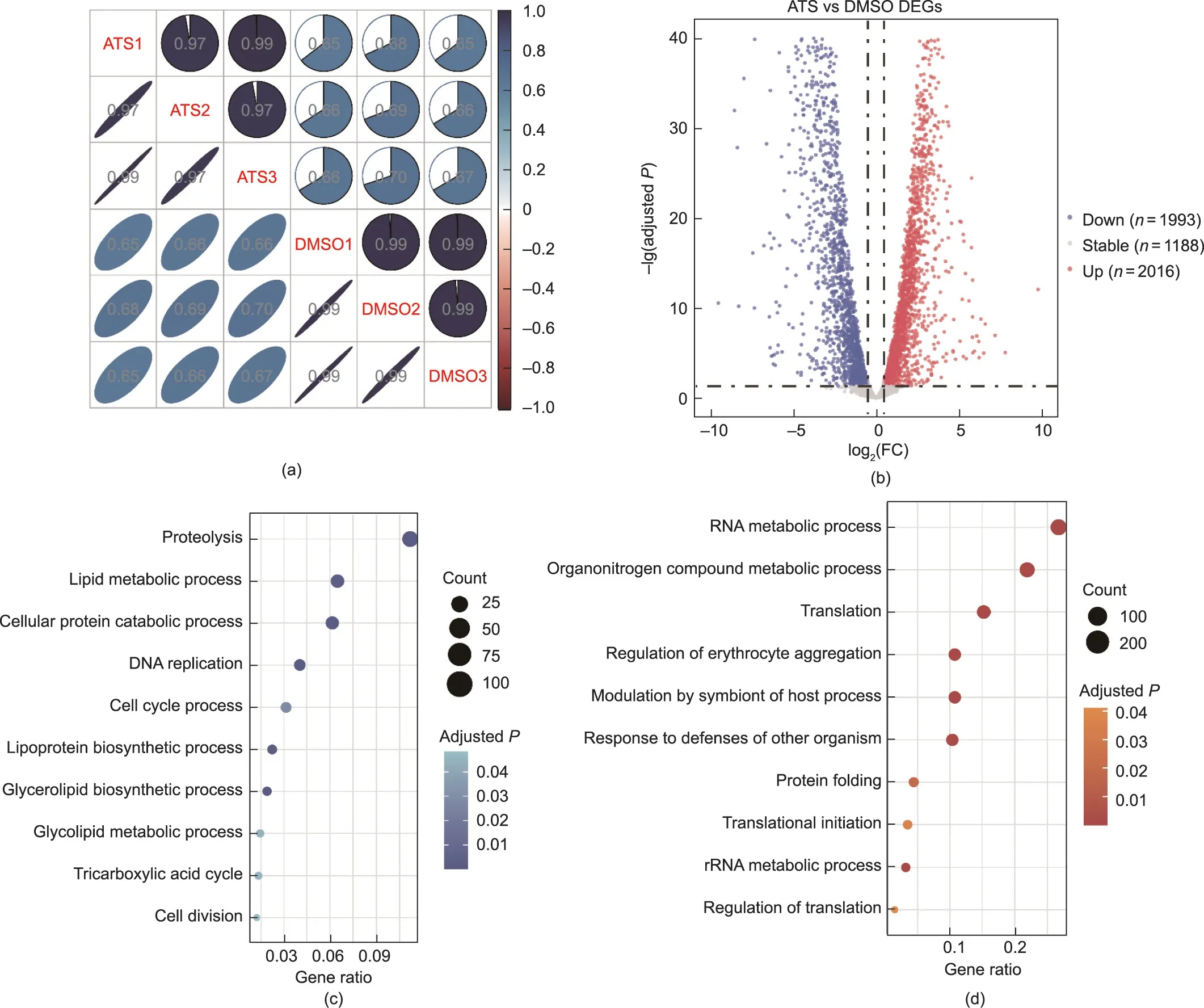
Fig.2.RNA-seq analysis of P.falciparum after treatment with ATS.(a)Correlation plot displaying the Spearman correlation coefficient between samples.Samples in the same treatment group were significantly correlated.(b)Volcano plot representing DEGs(P <0.05,log2(FC)>0.5)of P.falciparum after treatment with ATS versus the DMSO control.(c) GO-based enrichment analysis of downregulated DEGs.(d) GO-based enrichment analysis of upregulated DEGs.rRNA: ribosomal RNA.
The replication of Plasmodium parasites requires large supplies of nutrients and precursors from the host,including phospholipids,which are critical for the synthesis of cell membranes.The two most abundant phospholipid classes are phosphatidylcholine (PC)and phosphatidylethanolamine (PE), accounting for 75%-85% of the total phospholipids [32].As in many species, P.falciparum can use the de novo cytidine 5-diphosphocholine (CDP)-Cho and cytidine 5-diphosphoethanolamine(CDP-Etn)pathways to synthesize PC and PE, respectively, which is also known as the Kennedy pathway(Fig.5(a))[33].PC could be alternatively synthesized from serine and ethanolamine (Etn), which involves the transmethylation reaction of phosphoethanolamine methyltransferase (PMT)(Fig.5(a)).This alternative pathway is typically referred as the serine-decarboxylase-phosphoethanolamine-methyltransferase(SDPM) pathway [34].Notably, this SDPM pathway is absent in mammals; as a result, the relevant enzymes in the pathway are ideal targets for antimalarial intervention [35-37].
Here, we identified P.falciparum PMT(PfPMT,PF3D7_1343000)and P.falciparum ethanolamine kinase (PfEK, PF3D7_1124600) as potential target proteins of ART.PfPMT is expressed throughout the intraerythrocytic developmental cycle (IDC) and gametophyte and sporophyte stages of P.falciparum [38] and catalyzes the methylation of phosphoethanolamine (PEtn) into phosphocholine(PCho)[35].PfEK is the key enzyme that catalyzes the phosphorylation of Etn into PEtn, which serves as a substrate for the biosynthesis of PE and PC (Fig.5(a)) [20].
Similar to the validation experiments performed with Pf1-CysPxn, we first successfully expressed and purified both the PfPMT and PfEK proteins and then verified their specific binding to ATS(Figs.5(b)-(d)).The competition experiment suggested that cysteines are the key binding sites (Fig.5(e)).To confirm that ATS targets PfPMT and PfEK, we then carried out a lipidomic analysis.Significant changes in multiple lipid species were observed after ATS treatment,with 56 upregulated and 55 downregulated species(Fig.5(f);Figs.S4 and S5 in Appendix A).Among these,we noticed an apparent decrease in the abundance of many PC species and an increase in the PE content (Fig.5(g)) [32], indicating that the ATS indeed affects the lipid composition of parasites likely by targeting PfEK and PfPMT.As mentioned above, in the alternative SDPM pathway, Etn is first phosphorylated to PEtn by PfEK and then methylated to PCho by PfPMT[39,40].Previous research has shown that knockout of the PfPMT gene completely abolishes the biosynthesis of PC via the SDPM pathway in P.falciparum, leading to defects in growth and multiplication [41].Therefore, ATS may interact with PfEK and PfPMT to reduce PC synthesis,thereby interfering with the growth and reproduction of parasites.In addition,the increase in PE content could be explained by the inhibition of PfPMT function, which blocks the channel of PEtn methylation to PCho and thereby increases PEtn (which was unfortunately not measured in our study).
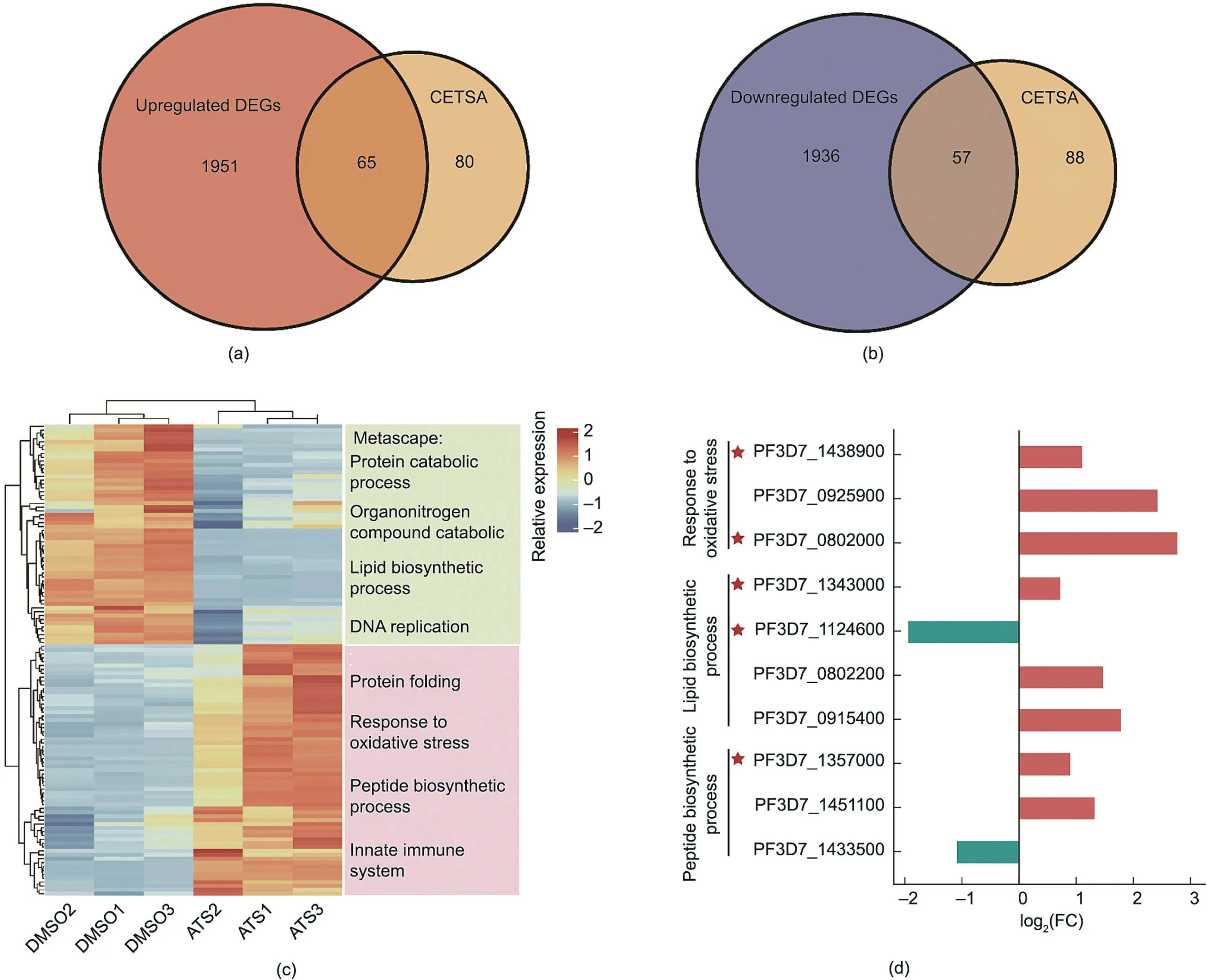
Fig.3.Comparative analysis of DEGs and MS-CETSA hit proteins.(a, b) Venn diagrams showing the overlap of upregulated and downregulated DEGs with target proteins derived from ITDR-MS-CETSA.(c)Heatmap representation of the mRNA expression levels of target proteins and the enriched biological keywords/pathways from Metascape.(d) Changes in expression level of the target proteins from three key biological pathways.
Therefore, ART may inhibit the catalytic activity of these two key enzymes, thereby affecting the synthesis of phospholipids(mainly PC species), ultimately leading to the inhibition/killing of malaria parasites [37].Nevertheless, further exploration is needed to fully determine the exact effects of ATS on the lipid metabolic processes of parasites in future studies.
3.5.ART inhibits the synthesis of parasite proteins
During the intraerythrocytic stage,the processes of transcription and translation are hyperactive in parasites because massive amounts of proteins need to be synthesized for development and proliferation [42].P.falciparum elongation factor 1-α (PfEGF1-α,PF3D7_1357000), a ubiquitously expressed protein during IDC,plays an indispensable role in translation and protein biosynthesis[42].Notably,there are two identical copies of the EGF1-α encoding gene (PF3D7_1357000 and PF3D7_1357100) in the P.falciparum genome [42].As shown previously, we identified PfEGF1-α as a potential target protein of ATS(Fig.3(d)).In addition,many ribosomal proteins showed thermal shifts in the CETSA assay,prompting us to explore whether ATS interferes with the translation process of the malaria parasite and the synthesis of new proteins.We first confirmed that ATS could specifically bind to PfEGF1-α through a series of in vitro and in vivo experiments similar to those performed in Figs.4(d)-(f), (h)-(j), and (m), including fluorescence labeling,competitive labeling, pull-down Western blotting, and confocal imaging (Figs.6(a)-(g)).Next, we used L-AHA, a nonradioactive L-methionine analog that can be incorporated into proteins during de novo protein synthesis,to monitor the effect of ATS on the synthesis of nascent proteins [43].Through the azide moiety on AHA,subsequent click chemistry reaction can occur with a fluorophore-alkyne probe and be visualized on an SDS-PAGE gel.AHA showed minimal toxicity to the parasites up to 100 μmol·L-1(Fig.6(h)).After the labeling conditions for AHA in parasites were optimized (Figs.6(i) and (j)), we showed that ATS could affect the de novo synthesis of parasite proteins(Fig.6(k)).

Fig.4.Validation of the targeting of ATS to rPfTrx-Px1(PF3D7_1438900)and rPf1-CysPxn(PF3D7_0802200).(a,b)Fluorescence labeling of rPfTrx-Px1 with AP1 in a(a)doseand (b) time-dependent manner.(c) Preincubation with excess ATS and IAA competes with AP1 binding to rPfTrx-Px1 proteins.(d-f) Fluorescence labeling of rPf1-CysPxn similar to the experiments performed on rPfTrx-Px1 in (a-c).(g)Fluorescence labeling of rPf1-CysPxn and rPfTrx-Px1 by AP1 under the same conditions.(h) Representative image of immunofluorescence staining of the colocalization of AP1 with rPf1-CysPxn proteins and (i) quantitative analysis of colocalization.(j) Pull-down Western blotting validation of the binding of AP1 to the Pf1-CysPxn proteins in situ.(k)ATS inhibits the enzymatic activities of rPf1-CysPxn.(l,m)ATS competes with the binding of the IAA-P to rPfTrx-Px1 and rPf1-CysPxn proteins.(n, o) Identification of the binding sites of ATS on the recombinant rPf1-CysPxn protein.ATS may bind to the cysteine 47 (C47) and phenylalanine 9(F9)sites of the rPf1-CysPxn protein.(p)Fluorescence labeling of AP1 with the wild type(WT),single-site mutants(F9A,C47A),and double-site mutant(F9A/C47A) of rPf1-CysPxn.(q) Enzymatic activities of the recombinant WT and mutant rPf1-CysPxn proteins (ns: not significant, *P <0.05, **P <0.01, ***P <0.001).Flu:fluorescence; CBB: Coomassie Brilliant Blue; DAPI: 4′,6-diamidino-2-phenylindole; m/z: mass-to-charge ratio.
4.Conclusions
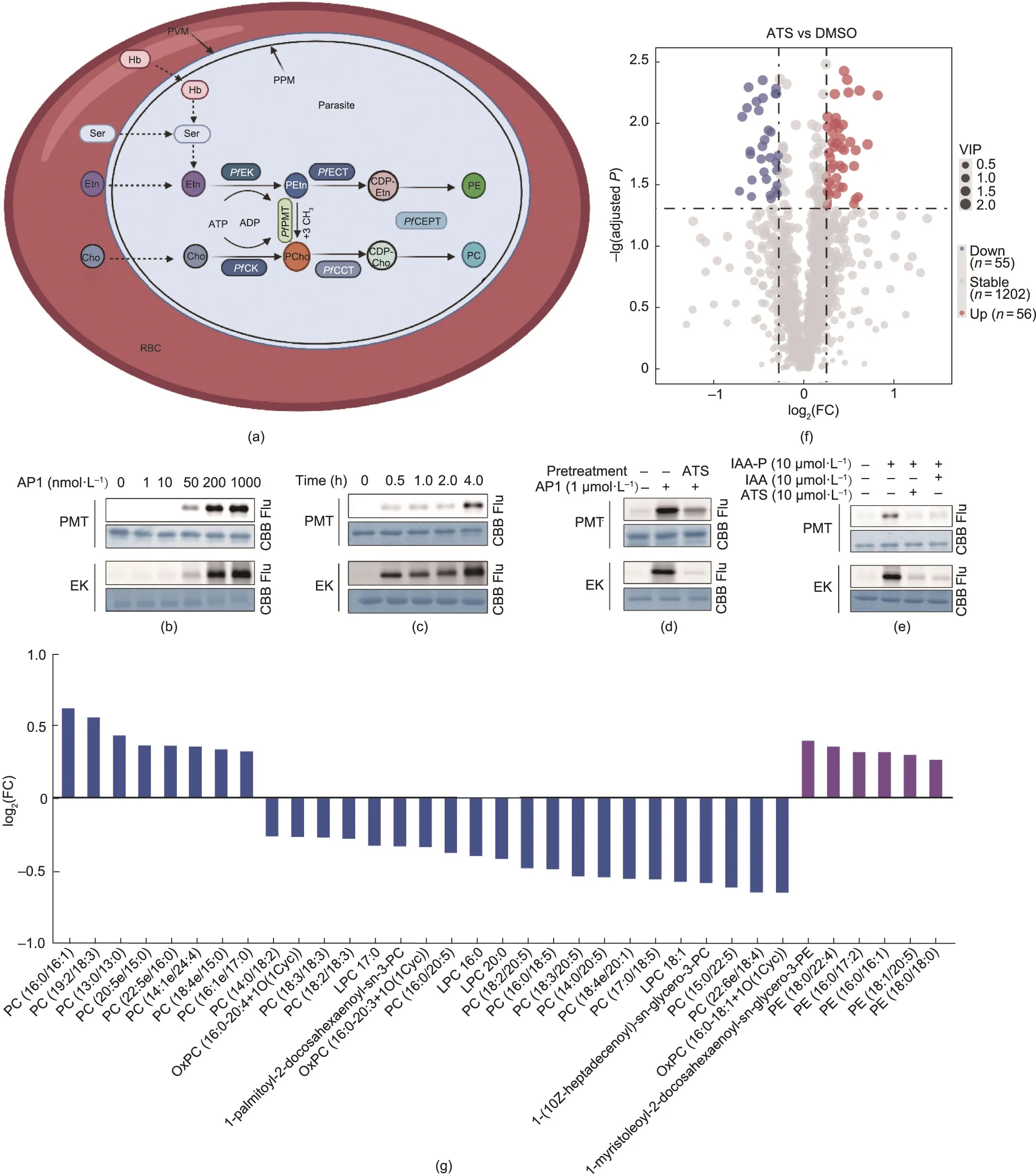
Fig.5.Validation of targeting of ATS to P.falciparum PMT(PfPMT,PF3D7_1343000)and P.falciparum ethanolamine kinase(PfEK,PF3D7_1124600).(a)Biosynthesis pathways of PE and PC in P.falciparum.(b,c)Fluorescence labeling of rPfPMT and rPfEK proteins with AP1 in a(b)dose-and(c)time-dependent manner.(d)Preincubation with excess ATS and IAA competes with AP1 binding to rPfPMT and rPfEK proteins.(e)ATS competes with the binding of the IAA-P to rPfPMT and rPfEK proteins.(f)Volcano plot showing differential lipid species with variable VIP >1,P <0.05,log2(FC)>1.2.(g)Bar plot showing the log2(FC)of significantly changed PC and PE subclasses,colored blue and purple,respectively.See Fig.S5 and Table S3 in Appendix A for details.PfCCT: P.falciparum choline-phosphate cytidyltransferase; Cho: choline; PfCEPT: P.falciparum choline/ethanolamine phosphotransferase; PfECT: P.falciparum ethanolamine-phosphate cytidytransferase; Etn: ethanolamine; Hb: hemoglobin; PCho: phosphocholine;PEtn: phosphoethanolamine; PPM: parasite plasma membrane; PVM: parasitophorous vacuolar membrane; RBC: red blood cell; Ser: serine; ATP: adenosine triphosphate;ADP: adenosine diphosphate; PfCK: P.falciparum choline kinase; OxPC: oxidized phosphatidylcholines; LPC: lysophosphatidylcholine.

Fig.7.ATS simultaneously interferes with ① redox homeostasis, ② lipid metabolism, and ③protein synthesis in P.falciparum to exert antimalarial effects by targeting several essential proteins.DV:digestive vacuole;HZ:hemozoin;Mito:mitochondria.
ART and its derivatives currently serve as the cornerstone drugs of malaria therapy,and the need for greater knowledge of its antimalarial mechanism has become increasingly urgent due to the emergence and spread of ART resistance.As it is known that ART can bind to proteins through different binding modes [44,45], we used MS-CETSA to identify the potential antimalarial targets of ART based on the principle that drug binding events affect the thermal stability of the targeting proteins.We then combined the MSCETSA results with transcriptomic analysis to determine the critical antimalarial target proteins and corresponding causal pathways.Subsequently, we carried out a series of verification experiments on four selected target proteins.Overall, this study indicated that ATS could bind to several critical proteins to interfere with the protein synthesis,lipid metabolism,and redox homeostasis of P.falciparum and exert its antimalarial effects (Fig.7).Our results help clarify the antimalarial mechanism of ART and drug resistance [46] and establish potential methodologies for mechanistic studies of other antimalarial drugs.Most importantly,the identification of critical targets and pathways serves to indicate new directions for the rational optimization of existing ACTs and the alleviation of ART resistance in parallel with other temporizing strategies [47].
Acknowledgments
The work was supported by grants from the National Key Research and Development Program of China (2020YFA0908000 and 2022YFC2303600); the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (ZYYCXTD-C-202002); the National Natural Science Foundation of China (82141001, 82274182, 82074098, 82003814,and 82173914); the China Academy of Chinese Medical Sciences(CACMS) Innovation Fund (CI2021A05104 and CI2021A05101);the Distinguished Expert Project of Sichuan Province Tianfu Scholar(CW202002);the Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences (CI2021B014); the China Postdoctoral Science Foundation(2022M721541);the Establishment of Sino-Austria‘‘Belt and Road”Joint Laboratory on Traditional Chinese Medicine for Severe Infectious Diseases and Joint Research (2020YFE0205100); the Excellent Scientific and Technological Innovation Training Program of Shenzhen(RCYX20210706092040048); the Fundamental Research Funds for the Central Public Welfare Research Institutes (ZZ14-YQ-051,ZZ14-YQ-052, ZZ14-FL-002, ZZ14-YQ-050, ZZ14-ND-010, and ZZ15-ND-10); the Introduce Innovative Team Projects of Jinan(202228029).
Compliance with ethics guidelines
Peng Gao,Jianyou Wang,Jiayun Chen,Liwei Gu,Chen Wang,Liting Xu, Yin Kwan Wong, Huimin Zhang, Chengchao Xu, Lingyun Dai,and Jigang Wang declare that they have no conflict of interest.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2023.06.001.
杂志排行

Engineering的其它文章
- Global Top Ten Engineering Achivements 2023
- 2023 Global Engineering Fronts
- Will Massive Appetite for Minerals Stall Clean Energy Transition?
- Optical Microscopy Advances Reach Sub-Nanometer Resolution
- International Correlation Research Program: Cross-Fault Measurement for Earthquake Prediction
- A Systematic Perspective on Communication Innovations Toward 6G