高通量计算与机器学习驱动高熵合金的研究进展
2023-03-22解树一尹海清刘斌斌曲选辉
张 聪,刘 杰,解树一,徐 斌,尹海清*,刘斌斌,曲选辉
(1 北京科技大学 北京材料基因工程高精尖创新中心,北京 100083;2 北京科技大学 钢铁共性技术协同创新中心,北京 100083;3 北京科技大学 新金属材料国家重点实验室,北京 100083;4 北京科技大学 新材料技术研究院,北京100083)
金属材料有着悠久的历史和广泛的应用,通常传统金属材料的设计方法一直都是以一种或两种金属元素为主元,通过添加少量的合金化元素来改善组织结构及性能[1-2]。高熵合金[3-4](high-entropy alloys, HEAs)至少含有四个主要元素,其元素原子分数通常在5%~35%之间[5-8]。起初对于高熵合金的研究大多是多相合金,而不是单相固溶体。近年来,随着对单相高熵合金的研究逐渐深入,在无其他相混杂影响下揭示了高熵合金的基本机制[5]。目前单相高熵合金的研究主要包括以Cantor合金为代表的FCC型单相体系[4]和以Senkov合金为代表的BCC型单相体系[9]。针对高熵合金单相研究而言,合金元素的数量、类型和浓度等变量可以系统地变化,并与合金物理力学性能直接相关,这对理解某些多相合金有更广泛的适用性。高熵合金如今发展到包括金属间化合物、纳米沉淀、陶瓷化合物以及只有三种主要元素的非等原子比高熵/中熵合金材料[10-12]。为了追求更好的力学性能,高熵合金的成分空间正在迅速拓展,例如曾报道过的具有超高强度和良好低温韧性的Al0.5Cr0.9FeNi2.5V0.2高熵合金[13]。针对高熵合金成分设计而言,当前可开发的成分空间巨大,因此如何更有效地选择最合适的成分以获得最理想的相结构和性能是一个重大挑战。
在寻找新合金的过程中,实验一直都是最有效的方法之一。为了克服传统“试错法”的偶然性,提高合金开发效率,高通量制备技术得以出现,其核心是设计并制备成分梯度以实现一批合金成分的高通量表征,例如扩散耦、超重力场和激光增材制造都有助于形成具有成分梯度的块体合金,或者使用多靶溅射来制备成分梯度薄膜[14-17]。
材料基因工程中的“高通量”思想可以加速高熵合金成分筛选,且有效促进高熵合金的开发。通常在追求目标性能时可能会存在元素成分或者种类偏离高熵合金规则的现象,若非规则外的性能优于静态合金设计规则的性能,则可认为这种现象是合理的[18]。对于高熵合金不断增长的成分空间,不仅增加了实验探索难度,而且对计算提出了更高的要求,也同时为机器学习的应用带来了机遇[19]。针对高熵合金工艺设计而言,采用机械设计的原则将传统合金力学机制转化应用于高熵合金是一种可行的手段[18]。在整个设计过程中,保持高熵合金特征优势的同时,将其与描述机制结合起来,极大地促进了高熵合金变体的开发研究[20-21],例如孪生诱发塑性(twinning induced plasticity, TWIP)高熵合金[20,22-24]、间隙高熵合金[25-27]、相变诱发塑性(tranformation induced plasticity, TRIP)高熵合金[28-30]以及多相高熵合金[31-32]。
激光增材制造技术有着良好的应用前景[33],基于高通量思想的增材制造技术可以大大缩短高熵合金开发周期[34-36]。与传统加工方法相比,增材制造在制备时自下而上逐层增加,突破了传统方法的限制,能够制备结构复杂的精密器件,且在制备大规模金属部件上也有着显著优势[37]。高熵合金具有高强度、优异的高温性能、良好的耐腐蚀性和耐磨性等优越性质,而使用传统铸造方法在工业上生产块状高熵合金存在固有的高精度工艺复杂性。增材制造有助于对局部过程进行高水平的控制,能够生产出复杂的几何形状,适合用于高熵合金的制备[38]。
目前应用在高熵合金领域的增材制造技术主要包括激光选区熔化(selective laser melting, SLM)[39]、激光熔融沉积(laser metal deposition, LMD)[40]和电子束选区熔化(electron beam melting, EBM)[41]。在现有的一些高熵合金中,基于增材制造技术进行的研究主要集中在以下体系[42]:CoCrFeNi系高熵合金、CoCrFeMnNi系高熵合金、AlCoCrFeNi系高熵合金、过渡族金属系难熔高熵合金等。其中增材制造难熔高熵合金(refractory high-entropy alloys, RHEAs)由于易产生成分偏析,一般采用高能量密度的小光斑激光器作为热源促进其元素均匀化,主要应用在制备具有一定优异性能(耐腐蚀性[43]、高硬度[44]、耐磨性[45-46]等)的高熵合金涂层方面。除此之外还可以根据层错能大小组合不同的缺陷进行微观结构设计[21,47]。针对高熵合金的性能设计,目前主要集中在高强度、强韧性[48-51]、抗氧化、耐磨性[52-56]等研究方面,制备并研究具有优越性能的目标合金。
近年来,理论和计算研究在材料科学中发挥着越来越重要的作用。计算机模拟和实验数据的结合有助于理解和阐释物理机制,从而能够有效地预测合金的性能。本工作主要介绍了三种方法,并提供了它们在高熵合金开发中的成功应用实例,包括高通量热力学计算、高通量第一性原理计算和机器学习。此外,还说明了不同高通量计算技术的优缺点和适用场景、存在的问题以及未来的展望,以便为高熵合金的开发设计提供指导。
1 基本概念
1.1 高通量计算
“高通量(high throughput)”一词在研究中广泛使用,2011年材料基因组计划(国内启动了相应的材料基因工程计划)提出后在材料领域广泛使用,并成为材料基因工程计算和实验区别于传统计算和实验表征的显著特征。材料领域的高通量计算(high throughput calculation,HTC)不同于计算机科学的高性能计算(high performance computing,HPC),前者旨在通过并行计算、高性能计算等手段,以提高计算通量的形式加速材料计算。高通量计算方法主要特征是并发式计算、自动流程计算,可以实现高数量级任务的高效计算而非多任务顺序自动计算。材料计算涉及多个尺度,高通量计算最早在量子尺度开展,充分满足了加速寻找新材料和优异性能的需求,尤其是新型功能材料和高性能材料的发现。高通量第一性原理计算方法在发现新材料、预测新材料性能或优化现有材料上应用较广。
高通量第一性原理计算可以解决材料成分的高效筛选问题,其经典案例是计算了80种二元合金的176种晶体结构并对其各性能进行了应用分级[57]。该思路用于多类材料的发现,其中包括高熵陶瓷材料的性能计算。国际上已经开发了基于高通量第一性原理计算的架构平台,如AFLOW[58]是包含352余万种化合物的7.3亿个性能数据的大型数据库。高通量第一性原理计算被认为是有潜力的快速材料筛选方法。
近年来高通量计算的概念被应用于微观热动力学尺度,成为金属结构材料快速获取相信息的方法[59]。高通量热动力学计算是近年发展起来的集热力学与动力学于一体的计算方法。基于热/动力数据库可以高效模拟任意数量组元间的热力学和动力学相变。对于扩散型相变模型可处理单相的均匀化、析出相的溶解和长大、第二相粒子的粗化以及凝固过程中的显微偏析等问题,从而实现材料成分和工艺设计。高通量第一性原理计算和高通量热动力学已在快速发现及筛选新材料方面广泛应用,包括工业铝合金筛选、高温合金组织设计、高熵合金成分优化等。
1.2 机器学习
数据科学作为理论研究、计算和实验外的科学发现“第四范式”,在2007年提出后即被各领域各学科广泛接受和应用。机器学习(machine learning, ML)是一个致力于理解和构建“学习”方法的研究领域,即利用数据提高某些任务集性能的方法[60]。随着以人工智能、大数据等为主导的“第四次工业革命”时代的到来,人工智能的新分支——机器学习已被广泛应用于机器人技术、计算机视觉、数据挖掘和生物医学等众多领域。机器学习是一门多领域交叉学科,涉及概率论、统计学、逼近论以及算法复杂度理论等[61]。
机器学习概念最早在1959年由Samuel提出[62],现已发展成为一个涉及计算机科学、统计学等多领域的交叉学科。机器学习因其高效的计算和预测能力,逐渐被用于材料科学研究领域。基于充分的实验研究和理论计算,机器学习方法可以快速完成数据挖掘,揭示其中所蕴含的信息和规律,并准确预测材料性能进而筛选目标材料。近年来,机器学习受到广泛关注,在材料科学开发新材料方面也展现出了卓越的能力。2016年发表在《自然》杂志封面的文章“Learning from failure”,挖掘大量失败实验数据成功实现化学反应预测和新化合物形成,这一案例进一步助推了机器学习在材料中应用的研究热潮[63]。
随着对材料基因工程思想的深入理解,高通量计算与机器学习在材料的成分设计筛选及性能预测与优化上得到大量应用。数据驱动方法可以明显加快研发速度,缩短研发时间,降低计算成本。不论是微观还是宏观层面,该方法都可以被应用于材料科学领域中的新材料发现、材料性能预测等[64]。
2 高通量计算及机器学习在高熵合金成分筛选和性能优化研究中的作用和效果
高熵合金[3-4]因其新颖的合金设计原理而引起了人们的极大兴趣。高熵合金尽管最初被定义为单相多元的等原子比或近等原子比的合金,但优异的性能不断被发现,所期望的合金性能有时会由于元素间的非直观相互作用而产生。图1归纳了Web of Science数据库中2013~2022年期间高熵合金相关热点词出现的次数以及高熵合金文章中一些关键词出现的频次(图1)。高熵合金的成分选择和制备涉及复杂的跨尺度物理化学现象,其原材料、合金成分和制备工艺等因素均会对微观组织和性能产生重要影响[1,65]。仅通过实验方法探索高熵合金组织及性能的作用规律较为耗时,而材料计算和机器学习技术被越来越多的人所接受和应用,成为高熵合金设计的有效研究手段[66]。
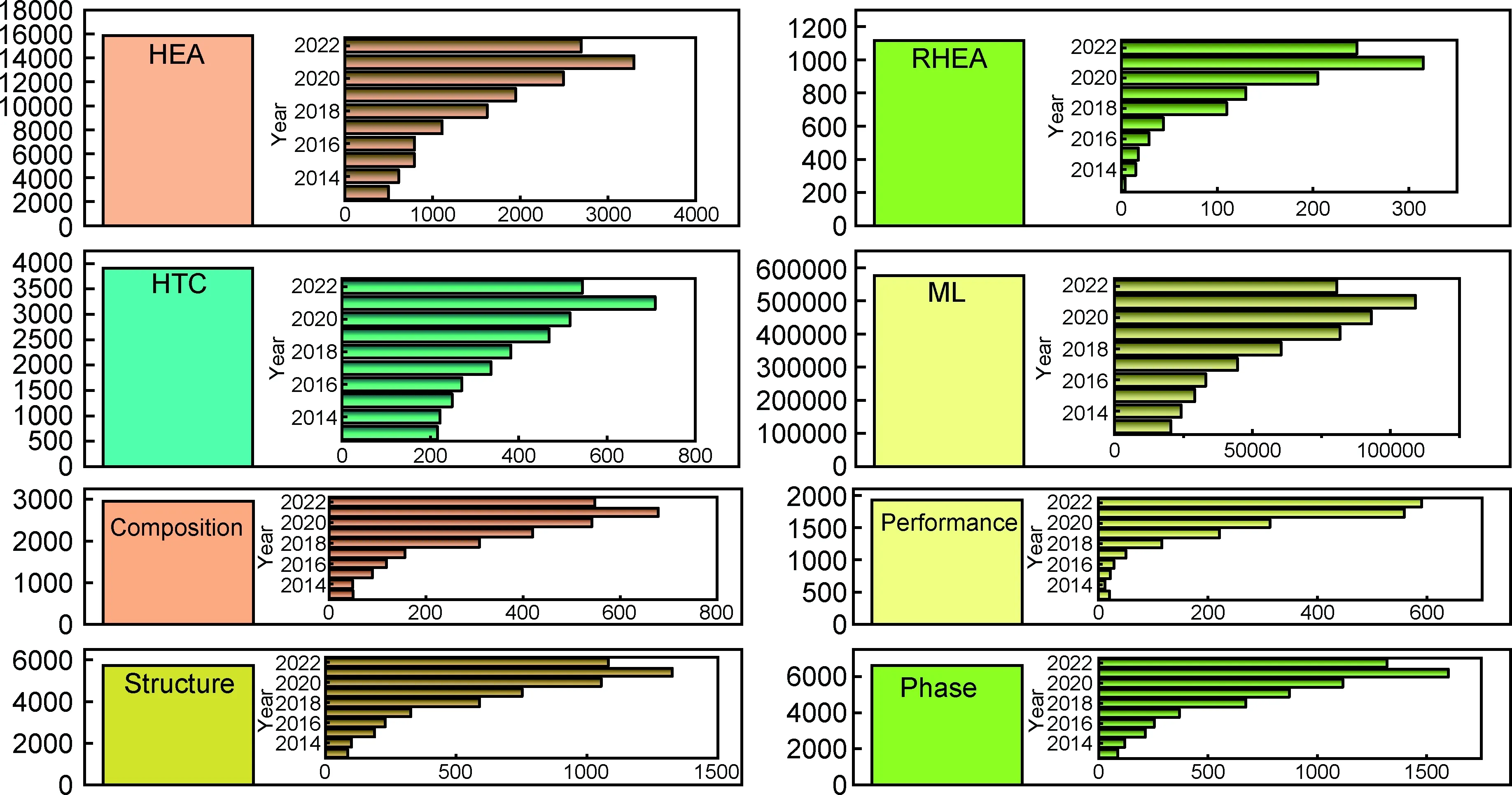
图1 Web of Science数据库中2013~2022年高熵合金领域相关热点词变化趋势Fig.1 Trend of hot words in the field of high-entropy alloys from 2013 to 2022 in the Web of Science database
2.1 高通量计算在高熵合金研究中的作用与优势
2.1.1 高通量第一性原理计算
随着超级计算机的发展,第一性原理的高通量计算作为一种成熟的计算方法研究材料性能,可以更加便捷和高效地解决复杂问题,对从微观角度理解和设计预测目标材料起到了不可或缺的作用。第一性原理计算基于量子力学计算凝聚态原子和电子的行为,定量描述原子键合的能力。第一性原理计算不依赖经验参数,通过输入元素种类和原子坐标,即可对材料性质数据进行预测,是理解材料本征、分析物理性质、设计新材料的重要工具。通过计算模型设计和参数优化,定量预测材料成分优选、物相组成、组织结构与性能的构效关系,实现高通量计算数据在新材料设计研发中的应用。基于第一性原理计算的高熵合金研究论文数量呈指数增长,证明了第一性原理计算方法的高计算精度可建立结构和特性之间的联系,为新材料的设计提供了充足的数据信息。高通量第一性原理计算具体作用与优势表现为三个方面:
(1)精准构建长程无序、短程有序结构。高熵合金是一种多主元混合合金,其结构特点为长程无序且短程有序,因此精准构建其原子排布结构是预测高熵合金材料特性的关键。目前已经开发的特殊准随机结构(special quasi-random structure,SQS)[67-68]与小集合有序结构(small set of ordered structures,SSOS)[69-70]能够反映高熵合金长程无序特性。第一性原理计算可以模拟高熵合金的平衡态结构,计算分析短程有序形成的影响因素,并且基于预选小集合有序结构(preselected small set of ordered structures, PSSOS)方法(图2[70])可以实现高熵合金的高通量第一性原理计算。因此,在构建多元组分长程无序、短程有序结构高熵合金方面,高通量第一性原理计算具有高精度优势。
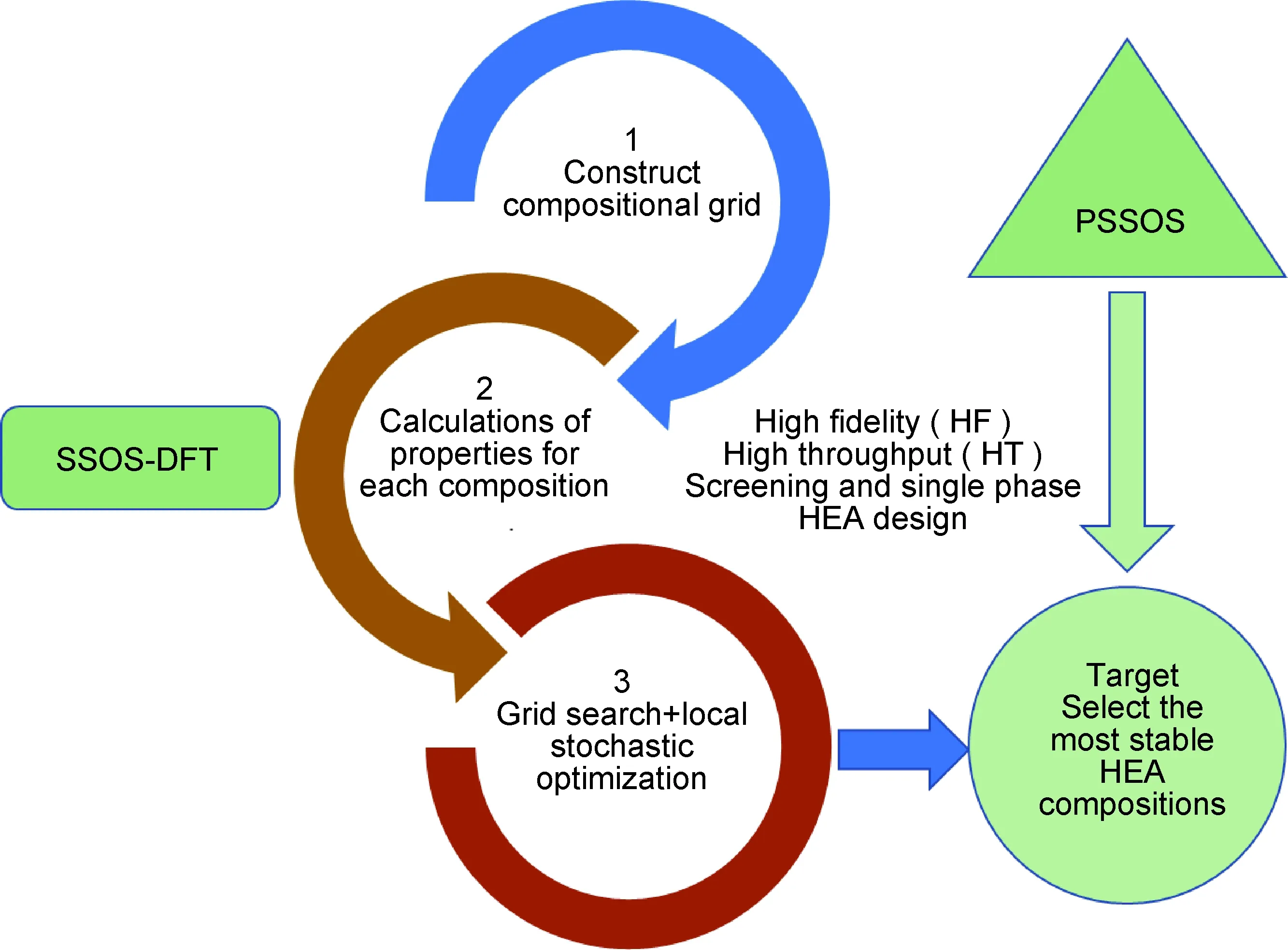
图2 高熵合金中基于SSOS方法的高通量第一性原理计算及筛选策略[70]Fig.2 Computational screening strategy based on SSOS high-throughput first-principle calculations method for high entropy alloys[70]
(2)准确预测高熵合金相稳定性。第一性原理计算可以获得合金原子级物理参数,判断合金的相稳定性以揭示合金元素添加对相稳定性的影响及合金分解中总能量的变化[71]。这种计算可以涉及任何系统,甚至是不存在的假设系统。随着高熵合金单一固溶体的形成,平衡状态可能包括多种固溶体和金属间沉淀物的形成。高通量第一性原理计算可确定单相的稳定性并预测单相中的元素偏析,这有助于深入理解难熔高熵合金的相结构性能[72]。从能量角度出发,基于FCC和BCC结构之间的晶格稳定性能量,高通量第一性原理计算可以筛选出具有BCC稳定性的候选合金。
(3)准确计算高熵合金的力学性能。具有单相晶体结构的高熵合金具有比强度高、延展性好及耐腐蚀性强等优异性能。第一性原理计算可得出单相体系弹性常数、剪切模量、体积模量和泊松比等参量,但当合金元素种类增加后可能会形成多相,其结构设计和性能预测则变得复杂得多。将量子力学的第一性原理计算与统计力学相结合,通过比较BCC相和第二相(如Laves相)对自由能的贡献来预测多相材料力学性能[73]。
2.1.2 高通量热动力学计算
高熵合金在制备过程中涉及复杂的热力学和动力学现象,材料的组织结构是其系统状态变量和制备工艺的产物,对性能有决定性作用,并对微观组织和性能产生重要影响[1-3]。高熵合金的性能很大程度上取决于其成分及微观组织,即组元数量、体系混合熵、体系混合焓、相体积分数、元素分布等,这些均可通过热动力学计算获得。高通量热动力学计算具体作用与优势表现为三个方面:
(1)精确获得高熵合金的相图及热动力学性质。热动力学计算方法基于相的吉布斯自由能和原子扩散系数来描述不同合金成分和制备工艺下多组元体系的相图和热动力学性质。较其他合金相比,热动力学计算高熵合金显得尤为重要,因为高熵合金已经不再受限于最初发现时的多元等原子比单相合金,还会出现双相或第三相,而性能也影响较大,甚至比单相时更佳。
(2)快速获得高熵合金关键组织参数。高通量热动力学计算可以快速获取广泛成分空间内合金的微观组织和热动力学性质,结合微观结构实验表征能够高效地研究组织结构的演变规律及其控制机理。掌握材料体系的热动力学信息不仅是研究材料微观组织演变的重要基础,也是开展不同尺度组织结构模拟(如扩散、相场、流场和有限元模拟)的必要条件。
(3)实现跨尺度分析。高熵合金制备过程中微观组织的定量描述是研发新型合金及提升现有合金性能的重要基础。结合微观组织的高通量热动力学计算并进行成分和工艺筛选是获得高性能高熵合金的有效途径。而高通量热动力学计算与高通量第一性原理计算相耦合,能够进一步加快合金开发中跨尺度集成计算的效率。在高熵合金研究中,基于高通量计算可以完成在包含百万、甚至千万个成分组合的超大成分空间中进行合金成分筛选与设计,实现从材料本征特性出发探索材料机理并研制新材料。
2.2 机器学习在高熵合金研究中的作用与优势
当考虑非等摩尔比的高熵合金时,研究者发现高熵合金拥有巨大的成分设计空间和自由度。而巨大的成分空间与对高熵合金理论认识的匮乏形成强烈反差,使得基于大数据的数据挖掘和机器学习方法成为除实验探索与计算模拟之外的有效方法之一,能够实现非等摩尔比高熵合金的设计。具体作用与优势表现为两个方面:
(1)高熵合金相形成的判定。基于体量较大但较为分散的数据集,以材料热动力学等参数或自定义参数为判据,机器学习可对数据进行分类以判定固溶体相的形成[74],这一类工作属于相对早期研究时采取的研究手段。随着研究的深入,发展了以合金的不同化学元素含量作为输入,或者以单质元素的物理、力学等本征特性的各类组合形式作为特征,如原子半径差、价电子数、构型熵、混合焓等,可直接建模得到元素组合与相形成的关系。例如基于人工神经网络算法建模获得相评估灵敏度矩阵,能够定量评估并调整设计参数实现形成固溶体、金属间化合物或非晶相的精准设计[75]。这为热动力学计算的相形成提供有力补充。
(2)高熵合金的成分与性能的关联关系。基于成分和元素的各类本征特性及其组合,以及基于材料理论知识[76]及特征工程思想对元素本征特征进行优化,可获得的综合反映合金不同特性的特征参量。通过构建特征参量与强度等性能的关联关系模型,可实现基于化学成分快速预测材料性能。该方法结合一定量的实验数据补充可为合金成分设计提供较好的指导作用[77],因此成为目前高熵合金成分设计的主要手段之一,尤其是对合金机理研究不足的情况下。机器学习技术在高熵合金上的成功应用,使得高熵合金在巨大成分空间的探索上能够兼顾可操作性与高效性。
3 研究与应用现状
3.1 高通量第一性原理计算方法在高熵合金的应用
(1)长程无序、短程有序分析。随着高熵合金微观结构研究的深入,高熵合金中原子的排布并非理想的无序状态,短程有序或多相共存现象是普遍存在的,同时也促进了第一性原理在高熵合金上的应用向更小尺度探索。在接近临界有序无序转变温度时,高熵合金的原子结构排列从完美的长程有序向局部的短程有序转变,而对于高熵合金短程有序(short-range ordering, SRO)结构的构建,将是高熵合金理论计算的基础和难点[78]。因此基于密度泛函理论(density functional theory, DFT)计算高熵合金的复杂性对模型构建方法提出了更高的要求。任县利等[79]提出了用蒙特卡洛结合密度泛函理论构建高熵合金平衡态结构的计算方法,分析短程有序形成的影响因素(原子间相互作用、原子间混合焓以及原子尺寸等)及短程有序的出现对高熵合金性质的影响,主要体现在电子分布、磁性以及力学性能方面。Nataraj等[80]采用第一性原理计算和团簇展开法考察了短程有序如何影响相稳定性和力学性能,其中团簇展开法用成对、三线态和高阶相互作用来表示结构的能量。通过几种线性和非线性算法对模型进行选择,确定了有效的团簇相互作用,进而研究高熵合金的相稳定性与温度的关系。
短程有序现象也与层错能有关,Tsuru等[81]结合蒙特卡洛模拟和第一性原理计算分析了Si元素的掺杂对FCC高熵合金性能的影响。研究表明Si的加入增加了局域晶格畸变,短程有序的形成引起了层错能的剧烈波动,并使其增加。低层错区和高层错区分布到基体中作为非均匀形核位置,这一特性导致了Si掺杂合金中超细孪晶的形成,被认为是提高合金强度和塑性的主要因素,使其在提高强度和延性方面具有很大的潜力[13]。对于高熵合金无序固溶体中存在短程有序的现象,第一性原理获得的晶体化学键信息可以作为评价高熵合金短程有序及各项性能的关键指标,如图3所示[82]。
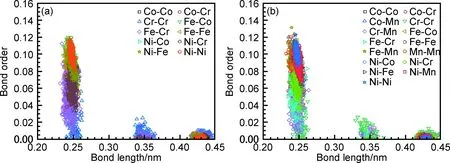
图3 高通量第一性原理计算所得合金的键序(a)和键长(b)[82]Fig.3 Bond order(a) and bond length(b) of alloy by high-throughput first-principles calculations[82]
(2)稳定相筛选。由于高熵合金成分空间广泛,目前尚无法稳定预测元素组合的单相形成能力。基于第一性原理计算提出了通过使用二元化合物形成焓的模型成为预测单相高熵合金体系的一种有效方法,如图4所示[83]。此第一性原理焓矩阵也可用于搜索单相合金的特定元素添加物,预测结果与已报道的实验结果表现出极好的一致性。
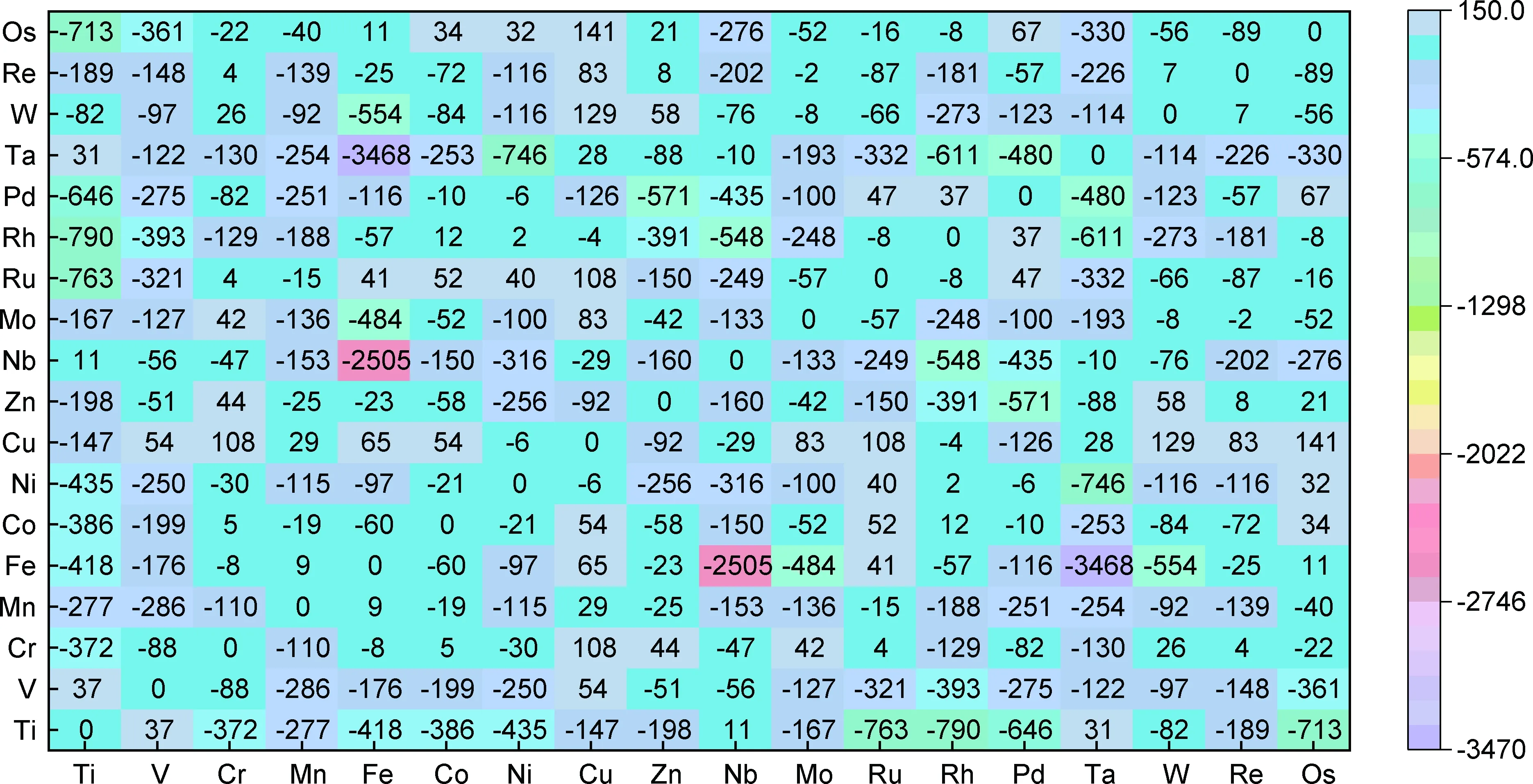
图4 二元化合物相对于分离成最低能量结构纯元素的形成焓[83]Fig.4 Formation enthalpy of lowest energy structure of binary compounds relative to phase separation into pure elements[83]
(3)计算高熵合金的力学性能。难熔高熵合金的相结构、体系能量和弹性模量直接影响高熵合金的力学性能。在固溶体中,层错能的大小影响部分位错的形成以及平面滑移和双交叉滑移的发生,它可以作为合金设计的可靠参数[84-86]。高通量第一性原理计算可以计算高熵合金的堆垛层错能(γusf)和表面能(γsurf),进而确定评估合金的强度和韧性。结合以BCC型难熔高熵合金γusf和γsurf为目标参量的统计回归替代模型,已通过VASP+SQS方法得到的计算值验证[87],证明高通量第一性原理计算在探究高熵合金强度塑性关系上具有优势。
单相晶系具有独立的弹性常数,因此第一性原理计算出的弹性常数需要根据现有的经验法则获得定量表达材料弹性性能的参数,如表1[88]所示。
通过计算原子尺寸差、价电子浓度和原子分布确定该合金主要结构为单一的无序BCC固溶体,采用第一性原理计算可获得合金化元素含量对体积模量、杨氏模量、剪切模量、韧脆转变指标B/G及泊松比的影响规律,进而可以确定最佳合金体系[89]。
3.2 热动力学计算在高熵合金中的应用
随着对高熵合金固溶体以及相变的研究,高熵合金热动力学计算数据库持续完善,热动力学计算在高熵合金研究中的应用也越来越多。
(1)计算高熵合金体系相组成。美国空军研究实验室[7,90]通过对数千种等摩尔成分高熵合金的高通量热动力学计算、筛选和分析,发现几乎所有单相等摩尔合金都是固溶体型合金,而大多数多相合金是固溶体与金属间化合物的混合型合金。计算结果与实验结果显示出良好的一致性[16,91]。美国田纳西大学[92]评估了固溶体相形成规律并确定这些经验规则适用于大多数合金成分,指出热动力学方法是一种有效的手段来预测高熵合金的相组成和相形成温度。
(2)快速筛选获得组织性能匹配的高熵合金体系。图5为基于高通量热力学计算筛选高熵合金的过程,首先利用平衡计算和非平衡希尔凝固计算相结合,基于高通量计算预测高熵合金加工后的熔点、相组成、热力学性质,快速获得满足熔点判据、相体积分数判据的合金成分空间,从而快速辅助分析有效的合金成分,降低实验试错频次[93]。
(3)辅助增材制造技术加工。通过高通量热力学计算结合多通道送粉增材制造技术,实现难熔高熵合金的高通量制备,利用离位X射线衍射(X-ray diffraction, XRD)和扫描电子显微镜(scanning electron microscopy, SEM)表征,获得各成分合金的相构成及元素分布,为MoNbTaW难熔高熵合金材料筛选提供关键信息[94]。
目前国内外已普遍接受采用高通量热动力学计算来筛选并优化高熵合金的成分和热处理工艺,对高熵合金广阔成分空间的探索有着重要的理论和实际意义。在筛选得到成分的基础上可开展高通量制备与表征,系统地掌握高熵合金组织及性能的演变机理,为高熵合金的开发设计奠定重要的理论基础。
3.3 机器学习在高熵合金中的应用
机器学习在近年来被广泛应用于高熵合金,利用机器学习方法进行材料性质预测和新材料设计的文章数量也呈现激增趋势。大数据时代的到来,得益于越来越多的开源数据库建立、算法不断改进以及计算机算力的飞速增长,基于数据库与机器学习的材料信息学已成为材料科学领域的重要研究手段。

表1 定量表达材料弹性性能的参数[88]Table 1 Parameters for quantitative expression of elastic properties of materials[88]

图5 高通量热力学计算筛选高熵合金[93]Fig.5 High-throughput thermodynamic calculation for screening high-entropy alloys[93]
(1)基于机器学习的固溶强化模型改进。机器学习特征筛选方法与高熵合金固溶强化模型结合,可以改进传统物理模型,其过程如图6[77]所示。通过构建高熵合金固溶强化的机器学习模型,发现电负性之差和剪切模量对高熵合金的固溶强化有显著影响,并能够构建比现有模型更加适用于高熵合金的硬度预测,同时可以进一步理解高熵合金元素电负性的差异驱动复杂原子环境中电荷的相互作用[95]。
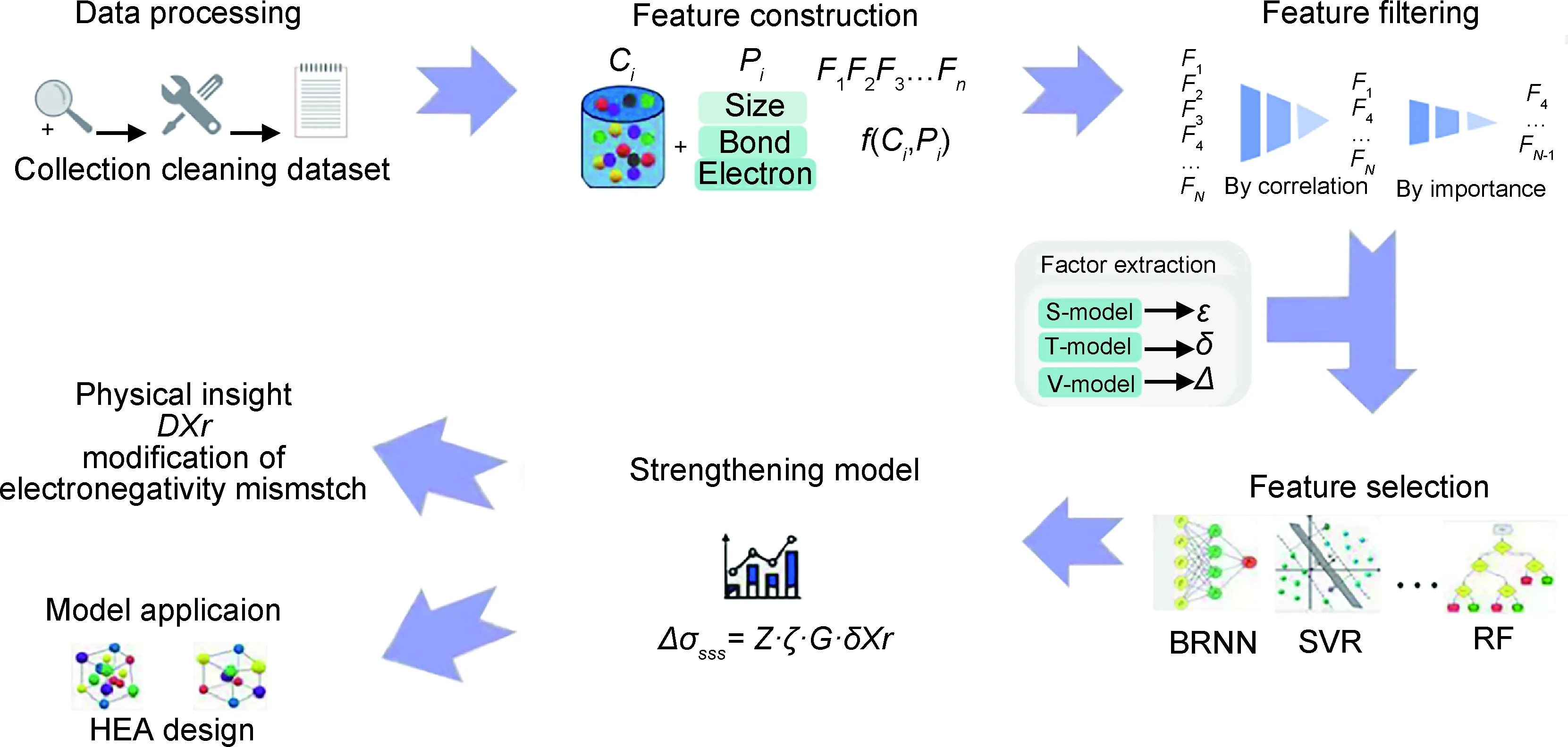
图6 固溶强化模型改进流程图[77]Fig.6 Flow chart of solution strengthening model improvement[77]
(2)高熵合金相种类预测。机器学习在相形成方面应用较多的方法主要包括神经网络(neural networks,NNs),高斯过程(Gaussian process,GP),K近邻(k-nearest neighbor,KNN),支持向量机(support vector machines,SVMs),主成分分析(principal component analysis,PCA),随机森林(random forests,RFs)和逻辑回归(logistic regression, LR)。这些研究表明价电子浓度、电负性、原子半径和混合焓都是决定相形成的重要特征。但是基于理想构型熵得到的混合熵相对较小,这可能是由于有限温度下的有序效应降低了材料的实际熵。基于晶系、晶胞尺寸以及形状结构参数并非主要因素,对于低溶解度/低浓度体系甚至适得其反。遗传算法在减少特征空间方面优于其他方法,如LASSO(least absolute shrinkage and selection operator)算法、随机森林和梯度提升决策树(gradient boosting decision tree, GBDT),而神经网络方法在固溶体形成类型方面具有优势。
高熵合金的相组成可分为固溶体相(SS)、金属间化合物(IM)以及混合相(SS+IM)。准确预测多组元高熵合金的相组成是筛选高熵合金性能的基础。根据高熵合金四大核心效应[96],使用机器学习算法建立了相预测模型可以高精度实现相分类,可以为合金设计提供指导[97]。
改进机器学习模型与材料描述符的组合可以进一步加快高熵合金的相分类。通过映射目标属性与材料描述符之间的关系,机器学习模型快速有效地构建材料描述符的最佳组合。Zhang等通过遗传算法系统框架从大量方案中有效选择最佳组合,该方法对固溶体和非固溶体的识别准确率高达88.7%,并进一步识别固溶体相中的BCC,FCC和BCC+FCC相,该遗传算法过程如图7所示[98]。
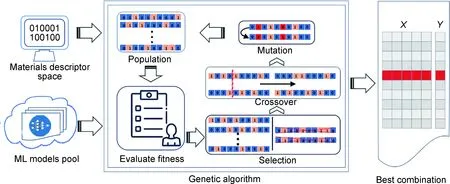
图7 遗传算法选择最佳模型与特征组合的步骤[98]Fig.7 Steps of genetic algorithm to select the optimized model and feature combination[98]
(3)机器学习模型预测高熵合金力学性能。在高熵合金的设计中,合金力学性能是至关重要的一部分。然而高熵合金存在原子相互作用复杂、原子环境和位错相互影响的问题[99]。Huang等在研究Cantor合金的硬度时使用了13个特征,包括第一电离能、价电子浓度等,显著多于相分类的5个特征,构建了优于传统理论硬度模型的高熵合金机器学习模型[100]。根据高精度硬度模型中特征对硬度的影响,在模型中引入电荷转移修正项,优化了传统经验公式预测精度,不同模型预测高熵合金硬度结果如图8所示。
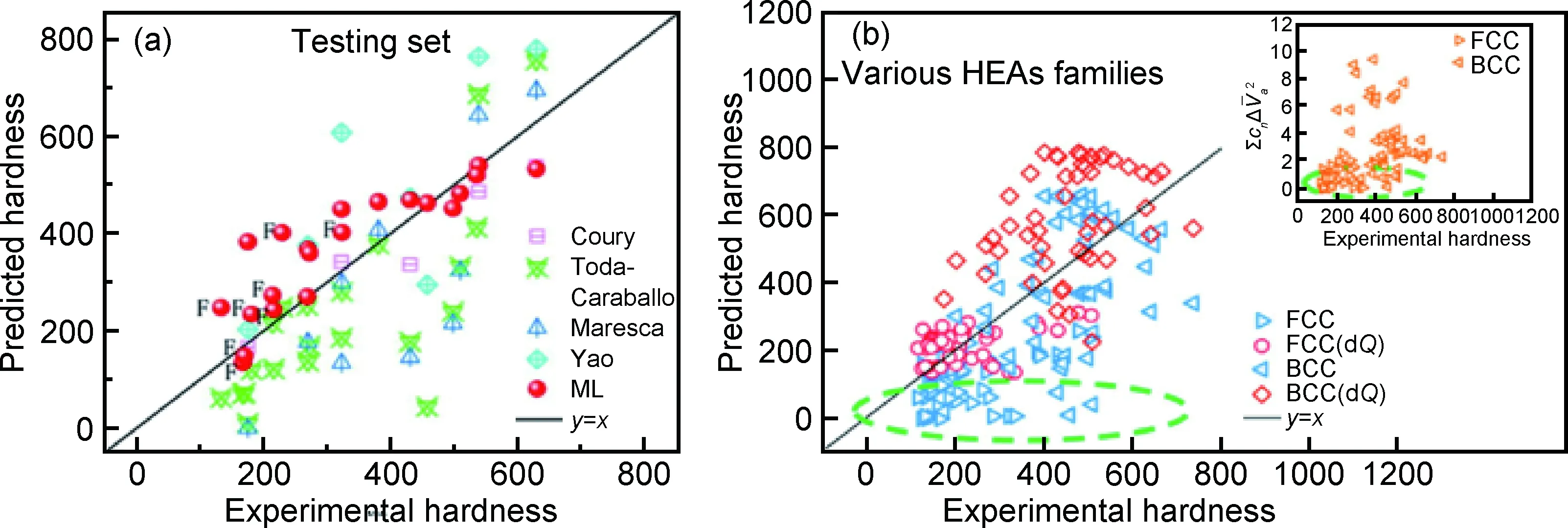
图8 机器学习模型与传统理论模型的硬度预测结果(a)以及理论硬度模型修正前后预测FCC,BCC合金硬度的对比(b)[100]Fig.8 Comparison of hardness predicted by ML model and the physical ones(a), as well as the hardness predicted by the modified physical models on FCC and BCC HEA alloys(b)[100]
(4)基于自适应设计的高熵合金优化。由于高熵合金的实验数据较少,用文献中收集的实验数据所构建的模型具有一定预测局限性,可采用自适应设计来提升模型精度以解决小数据机器学习建模问题。在高熵合金设计时采用一种以性能为导向的材料设计策略,结合机器学习、实验设计和实验反馈来寻找具有优异性能的高熵合金[101]。Rao等基于自适应设计思路在几乎无限的成分空间中加速新型高熵合金的设计(图9)[102]。利用该方法筛选并制备17种高熵合金,其中两种四主元高熵合金热膨胀系数与经典FeNi二元合金相当,两种五主元高熵合金的热膨胀系数比FeCoNiMnCu高熵合金低56.2%。
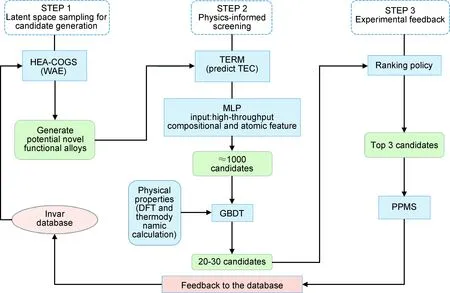
图9 基于机器学习模型、第一性原理计算、热力学模拟和实验反馈相结合的主动学习框架[102]Fig.9 Active learning framework based on combination of machine learning models,DFT calculations, thermodynamic calculations and experimental feedback[102]
该主动学习策略包括三个主要步骤:开发基于深度生成模型Wasserstein自动编码器体系的高熵合金生成方法HEA-COGS,结合无监督学习和随机抽样过程进行候选空间采样。采用包括多层感知器和梯度提升决策树的二阶段集成回归模型(TERM)进行物理信息筛选,建立基于高通量成分和原子特征的集成模型,在候选空间中推断出1000个可能的合金成分组合,再输入第一性原理和热力学计算的物理性质构建集成模型,将候选合金数量进一步缩小至20~30个。基于排名策略选择最有潜力的前三个候选合金,然后在物理性能测量系统(PPMS)中进行实验测量并反馈回数据库,重复迭代直到发现性能符合需求的目标合金。
4 存在的问题与未来发展方向
优异的性能赋予高熵合金许多潜在的应用前景,但是也存在一些问题会限制其发展。尽管有许多文献已报道过多种高熵合金力学性能方面的研究,但是对其变形机理的了解却很少,例如变形过程中的位错亚结构分析以及位错与溶质原子之间的相互作用机制等。
高熵合金主元的增加虽然使其拥有更大的成分空间,并引入更多的可能性,但是也给理论建模和仿真带来了重大挑战。主元数的增加使得在构建模型时需要考虑更多的化学相互作用,难以构建经验原子相互作用模型,若是使用传统的簇扩展方法或嵌入式原子法,则会导致过拟合。在进行第一性原理计算时,由于高熵合金结构的复杂性,需要使用超大的晶胞来构建结构模型,这会使计算成本以及时间大大增加。
虽然机器学习模型属于黑箱模型,其在作出预测的过程中进行的一系列操作是未知的、缺乏透明度且可解释性较差,但是机器学习仍可以帮助理解材料科学中的物理机制问题。例如,当使用可靠的机器学习模型与精心挑选的算法建模时,模型选择的描述符可能在变形机制中担任着重要的角色。同样可以使用机器学习来发现能够重现材料演变过程的新描述符,能够为构建理论模型提供参考。
尽管机器学习在高熵合金设计上具有优势,但是仍存在一些挑战。首先是模型置信度,机器学习预测的结果不能盲目地相信,需要对模型的不确定性进行量化,严格地估计每个预测的不确定性[103]。在实践中,有不同的方法来评估模型的不确定性[104](如图10所示),有DS理论[105]、模糊集[106]、区间方法[107]、概率方法[108]、贝叶斯方法等。通过了解和量化不确定性的来源,可以得出模型的置信区间,这不仅帮助理解预测的可靠性,也能通过在不确定性高的区域添加数据来实现主动学习[109]。
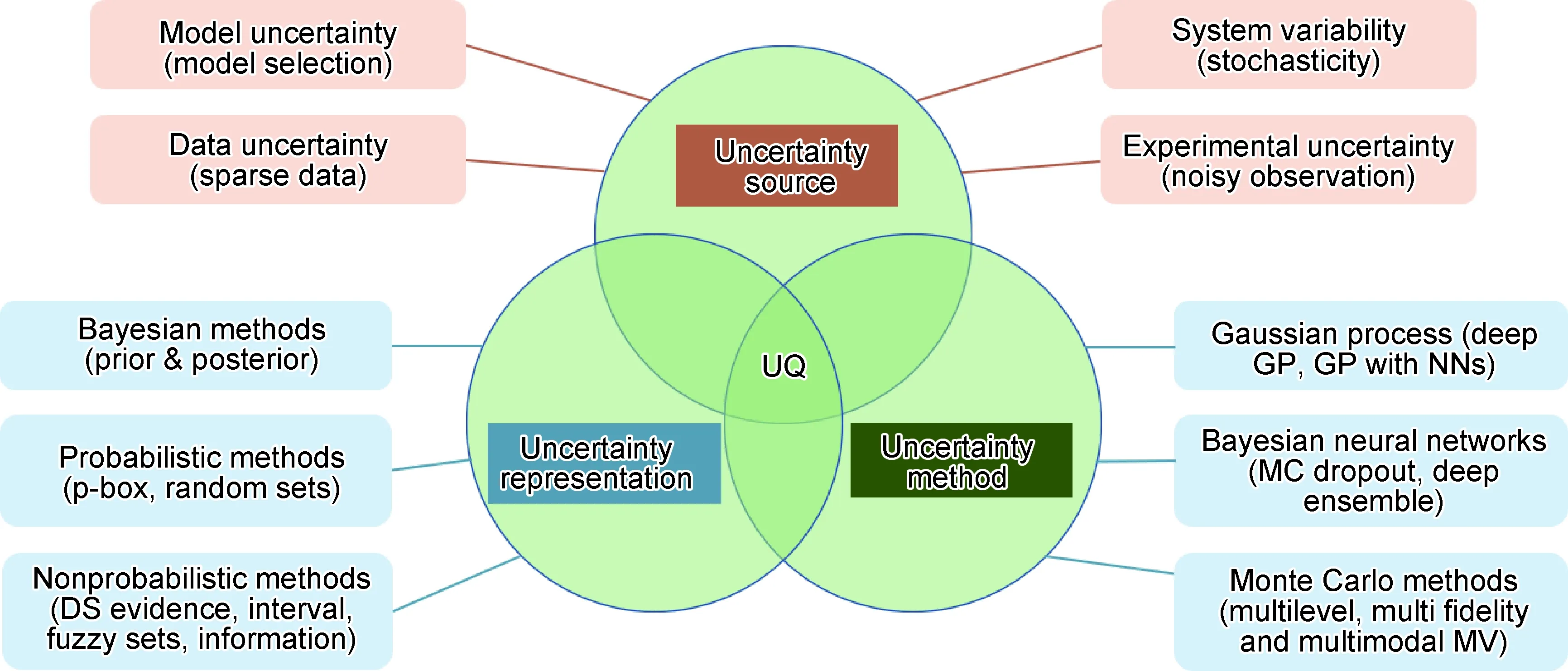
图10 不确定性量化UQ-ML模型中不确定性组成部分[104]Fig.10 Uncertainty quantification (UQ)-the components of UQ in ML models[104]
原子在材料中的排列具有各种对称性,如平移、旋转和晶体对称性,只有特定的点和线才能表示高维特征空间中的真实系统,保证原子的真实环境。目前主流的方法是构造一个保持对称性的描述符,但是相比之下,端到端的神经网络模型在自动捕获三维晶格上的物理相关描述符更具有优势,然而端到端的模型通常需要更大数据集来训练。获得高质量的数据集也是存在的问题,其难点在于不同的研究小组获得数据的实验条件不同,这使得在合并数据时需要仔细校准;此外,这些数据的获取比较耗时,在高熵合金问题上通常都只有小数据集能够使用。可以预见到,高质量的数据集依然是制约高熵合金机器学习发展的一大难题。
4.1 存在的问题
尽管近几年众多学者对高熵合金开展了研究,但目前对高熵合金的成分、工艺、微观组织与性能的复杂关系尚未完全揭示。总而言之,在材料计算和机器学习方面尚存在以下问题:
(1)第一性原理计算。由于包括多种合金组分和微观结构,第一性原理计算高熵合金往往比传统材料更加困难。而且,所需要的计算能力随着组元增加而不断增加。而一些传统的数据库对于高熵材料的计算远远不够,预测结果可能与实验结果差别较大。由于精度和计算力问题,第一性原理计算除了等原子比单相高熵合金之外,对非等原子比合金的探索工作应用较少。
(2)热力学计算。热力学计算的可靠性严重依赖于热力学数据库的准确度,目前商用高熵合金数据库在开发过程中仍存在计算精度低、部分体系相组成关系预测不准确等问题。同时,数据库的数据加密,用户无法自行增加和修正数据。国内缺乏可支撑高熵合金材料计算和设计的热力学数据库,严重制约着我国新材料的研发。
(3)机器学习。机器学习对高质量数据的依赖度较高。高熵合金数据的获取成本高且分布不均匀,易导致机器学习的过拟合问题,直接影响模型预测效果。另外,机器学习的可解释性差,难以说明因果关系的问题一直存在。无论在性能预测还是机理模型构建上,在研究时间尚短且积累数据有限的高熵合金上此问题可能更为突出。
4.2 未来发展方向
随着对高熵合金的研究深入和潜力应用的探索,第一性原理计算、热动力学计算、机器学习等作为材料发现与机理探究的先进工具,将在今后研究中充分发挥其效力。
(1)高精度的第一性原理计算加速方法。计算效率与精度的矛盾在高熵合金计算上显得更加突出,目前国际上支撑高通量第一性原理计算及机器学习技术与工具包括AiiDA和MatCloud等,都难以适应高熵合金的计算。因此,开发基于机理且兼具高通量与高精度的第一性原理加速器,将满足高熵合金研发提速的需求。
(2)适应高熵合金复杂性与数据稀缺问题的机器学习新方法。微软研究院的计算机专家与数据专家推出的AI for Science,将推动机器学习和人工智能与材料领域研究的深度合作。人工智能助力高熵合金机理模型、人工智能与高性能计算两大手段,有望突破高熵合金的复杂、高维、小数据的问题,也将形成今后高熵合金研究发展中的一个交叉学科分支。
(3)基于高通量计算与机器学习相融合的材料性能及服役行为全局优化。近年来我国材料的研究和生产强调全产业链管控,材料服役行为将逐步成为材料成分设计过程的约束条件之一,这使得研究过程进一步复杂。制备过程各工序的机理研究都涉及不同尺度,包括利用第一性原理和分子动力学方法对缺陷的研究,采用高通量跨尺度计算或机器学习辅助建模,机器学习实现各工序间的跨越,最终对材料性能与服役行为全局优化。
(4)高通量计算助力高熵合金的功能开发。未来高熵合金的研究与应用不局限于结构材料,功能材料同样存在巨大潜力。而高通量第一性原理在功能材料的构效关系研究上一直具有显著的优势,第一性原理计算探索高熵合金用作电池材料、磁性材料、核聚变/裂变材料等研究已开展,今后可能在新能源材料等亟需发展的材料领域上取得突破。
(5)助力未来高熵合金智能制造与数据库。智能制造是我国制造业提高产品质量的解决方案,而数据化转型是实现智能制造和降低人工劳动力的重要举措。高通量计算与机器学习是智能制造和数字化转型的落地实施手段,高质量数据是机器学习的基础。因此学习发达国家几十年高质量数据积累的经验,整合高通量计算、机器学习及实验数据,构建高质量高熵合金数据库,以期实现未来高熵合金数字化和智能化制造。
