过渡金属配位铝氢化物的研究进展
2023-03-22苏高琴曹志杰
苏高琴,马 宁,石 哲,曹志杰
(宁夏大学物理与电子电气工程学院,宁夏 银川 750021)
1 前 言
随着全球人口的快速增长和工业化程度的不断提高,能源需求量与日俱增,而传统的化石能源存量剧减、环境污染加剧等问题凸显,发展可再生清洁能源成为了人类社会可持续发展的必然要求[1, 2]。氢能作为高效可再生的绿色能源,其大规模发展和应用有望解决当前化石能源过度消耗和环境污染的双重危机,因而备受关注。氢能的规模化应用包括氢的制取、储运和高效利用三大技术环节,其中安全高效的存储技术是关键[3]。对于轻型车载燃料电池而言,美国能源部(DOE)提出了储氢系统的最大工作温度区间为-40~85 ℃,2025年的目标质量和体积储氢密度分别达到5.5%(质量分数,下同)和40 g/L,最终目标为6.5%和50 g/L[4]。氢气的储运方式主要包括气态储氢、液态储氢、固态储氢和有机液体储氢等。高压气态储氢是目前最常用的存储方式,如丰田燃料电池汽车Mirai采用70 MPa高压气态储氢罐[5],质量密度较高,但成本高且安全隐患大。低温液态储氢的质量和体积密度均非常高,但液氢需要冷却至-253 ℃,制冷过程能耗大,且液氢极易挥发,不利于长期使用。有机液体储氢的储氢密度较高,可用现有管道设施进行储运,安全方便,但操作技术条件较为苛刻,且易发生副反应,难以实现“零碳排放”目标[6]。固态储氢是指通过固态储氢材料进行可逆吸放氢,具有体积密度高、储运方便、安全性好等优点,是目前最具发展前景的储氢方式之一[7-10]。

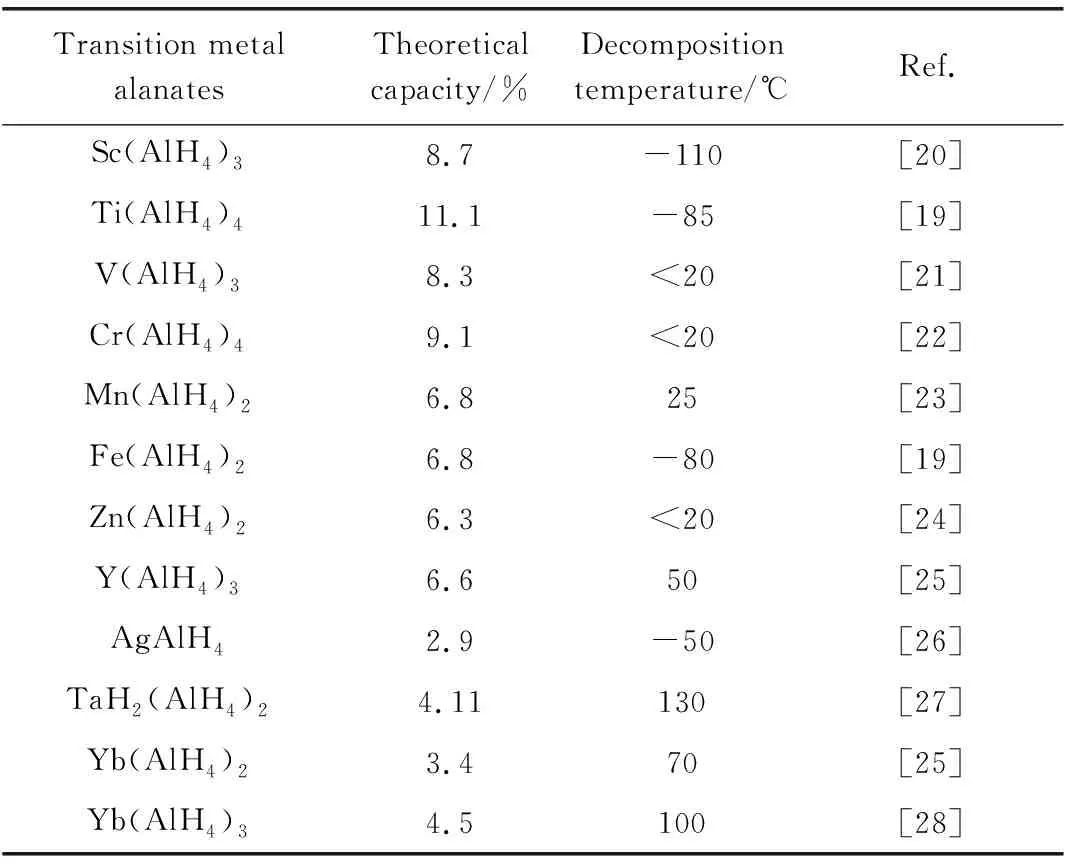
表1 过渡金属配位铝氢化物的物理化学性质
目前,常温甚至低温下的高容量固态储氢成为当前国内外储氢材料研究的难点之一。过渡金属配位铝氢化物具有储氢量高、热力学稳定性低、放氢温度低等特点,且热力学稳定性随着金属阳离子的变化可调范围较大,有可能满足低温固态储氢系统的应用要求。但当前,关于过渡金属配位铝氢化物的研究较为零散,多数设计与制备过程主要依靠经验规律,尚未得到完善的理论和技术,同时其储氢性能、分解特性及机理研究尚处于探索阶段。本文综述过渡金属配位铝氢化物的研究进展,主要包括制备方法、研究近况、未来的发展方向和挑战等,以期为过渡金属配位铝氢化物的深入系统研究提供参考。
2 过渡金属配位铝氢化物的制备方法
过渡金属配位铝氢化物的合成主要包括液相法和固相法2大类[29]。由于氢化物的热稳定性较低,为防止氢化物分解,其液相合成过程通常在低温下进行,而固相合成过程则在高压下进行。同时由于这些氢化物遇到氧或水时易燃,所有处理过程均需在保护气氛下进行。
2.1 液相法
液相法通常是通过金属卤化物与LiAlH4或NaAlH4在有机溶剂(如四氢呋喃、乙醚等)中发生复分解反应合成TM(AlH4)n[30]。以过渡金属氯化物TMCln和LiAlH4为例,其反应方程如下:
为了抑制过渡金属铝氢化物的分解,液相法通常在-80 ℃甚至更低的温度下进行,如Ti(AlH4)4和Fe(AlH4)2的合成温度低至-110 ℃[19]。合理调控反应过程中的温度、搅拌速度、反应时间等参数对于氢化物的合成至关重要。液相法能够灵活调整反应温度,对制备装置要求低,但反应时间较长,且有机溶剂会和氢化物形成较为稳定的络合物,导致释放的氢气不纯[20]。
2.2 固相法
固相法通常是通过机械球磨促进金属卤化物与LiAlH4或NaAlH4的复分解反应合成TM(AlH4)n,反应原理与液相法相同。
为防止氢化物在制备过程中发生分解,球磨过程通常在高压下进行,如Y(AlH4)3[31]和Yb(AlH4)3[28]的制备过程分别在8和10 MPa的氢压下进行。合理调控球磨过程中的球料比、压力、球磨转速等参数对过渡金属配位铝氢化物的合成至关重要。与液相法相比,固相法具有操作简便、反应时间短、不含有机溶剂等优点[32],但目标产物中存在卤化物,会降低体系的储氢量。同时,反应在高压下进行,需要耐压球磨装置和特殊的实验环境。
3 过渡金属配位铝氢化物的研究
3.1 Sc(AlH4)3
Sc(AlH4)3的理论储氢量为8.7%,到目前为止还未见实验上成功合成此种氢化物的报道。2008年,Charkin[21]从理论上提出了Sc(AlH4)3热分解的3种假设路径:
Sc(AlH4)3→HSc(AlH4)2+AlH3
Sc(AlH4)3→H2Sc(AlH4)+2AlH3
Sc(AlH4)3→ScH3+3AlH3
通过理论计算,3种分解路径的解离能分别为39.2,76.1和115.6 kcal/mol。
2017年,作者课题组[20]首次尝试采用固相和液相2种方法制备Sc(AlH4)3。在固相法制备过程中,即使在16.5 MPa的氢压下,Sc(AlH4)3在球磨过程中已经完全分解,说明Sc(AlH4)3在常温下非常不稳定。在液相法制备过程中,将LiAlH4和ScCl3的乙醚溶液冷却至-110 ℃进行反应,得到的产物是由Sc(AlH4)3和乙醚形成的络合物Sc(AlH4)3·n(C2H5)2O。该络合物能够在室温下保持稳定,说明有机基团能够增强Sc(AlH4)3的稳定性。然而,络合物在加热除去有机基团的过程中,氢化物同样会发生分解,反应如下:
3.2 Ti(AlH4)4
Ti(AlH4)4的理论储氢量为9.3%。1951年,Wiberg等[33]在-110 ℃下通过TiCl4和LiAlH4在醚溶液中反应,首次合成出淡黄绿色的Ti(AlH4)4,反应如下:
加热时,Ti(AlH4)4在-85 ℃以上开始分解放氢,颜色逐渐变深。达到室温时,放氢更剧烈,1~3 d后完全分解,放氢反应如下:
1975年,Kost等[19]采用同样的方法制备出Ti(AlH4)4,发现该化合物非常不稳定,且对空气和水极其敏感。与Wiberg等[33]认定的一步分解路径相比,Kost等[19]认为,Ti(AlH4)4在加热过程中的放氢反应分3步进行:
他们发现,用乙醚清洗并干燥后,Ti(AlH4)4在20 ℃下放置3~5 d会缓慢分解并释放60%~70%的氢。XRD结果表明,Ti(AlH4)4在20 ℃下处于非晶态,且热分解产物中存在金属Ti和Al。
3.3 V(AlH4)3和Cr(AlH4)4
V(AlH4)3和Cr(AlH4)4的理论储氢量分别为8.3%和9.1%,到目前为止还未见实验上成功合成这2种氢化物的报道。2008年,Charkin[21]从理论上提出V(AlH4)3热分解的3种假设路径:
V(AlH4)3→HV(AlH4)2+AlH3
V(AlH4)3→H2V(AlH4)+2AlH3
V(AlH4)3→VH3+3AlH3
通过计算可知,3种分解路径的解离能分别为43.9,85.0和120.1 kcal/mol。
2009年,Charkin[22]从理论上提出,Cr(AlH4)4热分解的假设路径,解离能为~0 kcal/mol,反应如下:
Cr(AlH4)4→HCr(AlH4)3+AlH3
3.4 Mn(AlH4)2
Mn(AlH4)2的理论储氢量为6.8%。1967年,Mackay等[23]通过MnCl2和LiAlH4在乙醚溶液中反应制备出Mn(AlH4)2,此氢化物在25 ℃下便开始分解放氢。到目前为止,未见关于Mn(AlH4)2结构和储氢性能的进一步报道。
3.5 Fe(AlH4)2
Fe(AlH4)2的理论储氢量为6.8%。1956年,Schaeffer等[34]首次研究发现,FeCl3和过量LiAlH4在乙醚溶液中-45 ℃下可发生反应生成Fe(AlH4)2。Fe(AlH4)2在室温下不稳定,能够分解形成较为稳定的中间氢化物FeAl2H6,并进一步分解放氢形成Fe和Al,具体反应如下:
FeAl2H6→Fe+2Al+3H2
与Schaeffer等[34]的结论不同,Neumaier等[35]认为FeCl3和LiAlH4在-116 ℃下乙醚溶液中反应会先形成不稳定氢化物Fe(AlH4)3,随后分解放氢生成Fe(AlH4)2。Fe(AlH4)2在室温下能够稳定存在,但在加热至200 ℃的过程中缓慢分解,具体反应过程如下:
FeCl3+3LiAlH4→Fe(AlH4)3+3LiCl
Fe(AlH4)3→Fe(AlH4)2+AlH3+1/2H2
Fe(AlH4)2→Fe+2Al+4H2
Kost等[19]则认为,Fe(AlH4)2在-80 ℃时会缓慢分解,达到20 ℃时放氢明显加快,反应如下:
Fe(AlH4)2→Fe+2AlH3+H2
但是,关于Fe(AlH4)2的形成过程和稳定性尚存在一定争议。
3.6 CuAlH4
CuAlH4的理论储氢量为4.2%。1952年,Wiberg等[36]研究发现,采用CuI和LiAlH4在室温下反应不会产生CuAlH4,而是直接生成LiI、AlI3和CuH,反应如下:
1977年,Ashby等[37]发现CuI和LiAlH4在四氢呋喃中-78 ℃下反应能够生成CuAlH4,但此氢化物非常不稳定,能够迅速发生分解,并与产物CuH反应生成Cu3AlH6,具体如下:
CuAlH4→CuH+AlH3
2CuH+CuAlH4→Cu3AlH6
3.7 Zn(AlH4)2
Zn(AlH4)2的理论储氢量为6.3%。1951年,Wiberg等[38]和Barbaras等[39]几乎同时提出,采用ZnI2和LiAlH4在室温下反应不会产生Zn(AlH4)2,而是直接生成LiI、AlI3和ZnH2,反应如下:
ZnI2+2LiAlH4→2LiI+2AlH3+ZnH2
Zhizhin等[24]认为,ZnI2和LiAlH4反应首先会形成Zn(AlH4)2,随后与LiAlH4反应进一步生成Lin[Zn-(AlH4)2+n],产物的成分和参与反应的LiAlH4的量有关,具体如下:
ZnI2+2LiAlH4→Zn(AlH4)2+2LiI
Zn(AlH4)2+nLiAlH4→Lin[Zn(AlH4)2+n]
到目前为止,未见关于Zn(AlH4)2的形成过程以及分解放氢特性的进一步报道。
3.8 Y(AlH4)3
Y(AlH4)3的理论储氢量为6.6%。1978年,Kost等[25]通过LiAlH4和Y的卤化物在乙醚溶液中-110 ℃下发生置换反应首次制备出白色的Y(AlH4)3。热力学研究表明,Y(AlH4)3的起始放氢温度为50 ℃,结合-110~25 ℃的放氢量,他们提出Y(AlH4)3的放氢反应如下:
2017年,作者研究组[31]首次采用高压球磨法成功制备出Y(AlH4)3,并系统研究了其放氢反应机制、放氢性能和可逆性。研究表明,Y(AlH4)3在产物中以非晶的形式存在(图1a),放氢过程分为4步进行(图1b)。根据放氢过程的物相变化(图1c),Y(AlH4)3的4步放氢反应如下:
其中,第一步放氢过程的起始放氢温度低至80 ℃,140 ℃时30 min内的放氢量高达3.4%,且放氢激活能低至91.7 kJ/mol。在145 ℃和10 MPa H2条件下,Y(AlH4)3第一步放氢过程的可逆储氢量达到了2.6%(图1d),为氢化物理论储氢量的75%。

图1 Y(AlH4)3-3LiCl放氢过程的物相分析及吸放氢曲线[31]: (a) 提纯前后的XRD图谱,(b) 升温放氢脱附(TPD)、质谱(MS)和差热分析(DSC)曲线,(c) 在不同温度下放氢产物的XRD图谱,(d) 在145 ℃、10 MPa H2条件下的吸放氢曲线Fig.1 The dehydrogenation phase analysis and isothermal ab/desorption curves of Y(AlH4)3-3LiCl[31]: (a) XRD patterns before and after purification, (b) TPD, MS and DSC curves, (c) XRD patterns of dehydrogenated samples at different stages, (d) isothermal absorption and desorption curves at 145 ℃ and 10 MPa H2
3.9 Zr(AlH4)4
Zr(AlH4)4的理论储氢量为7.5%。1957年,Reid等[40]在室温、氦气气氛保护下通过Zr(BH4)4和LiAlH4在醚溶液中反应首次制备出Zr(AlH4)4,反应如下:
白色的Zr(AlH4)4在室温下非常不稳定,在几个小时内便发生分解形成黑色固体。
2007年,Epshteyn等[41]在0 ℃下通过LiAlH4和ZrCl4在乙醚溶液中反应合成Zr(AlH4)4,反应路径如下:
此工作没有关于Zr(AlH4)4放氢特性及结构表征相关的报道。
3.10 Nb(AlH4)n及相关铝氢化物
1965年,Wiberg等[42]研究发现,NbCl5和过量LiAlH4在醚溶液中反应,能够生成Nb(AlH4)n和LimNb-(AlH4)n+m这2种铝氢化物。生成Nb(AlH4)n的反应如下:
NbCl5+5LiAlH4→
产物Nb(AlH4)n的成分与反应温度和参与反应LiAlH4的含量密切相关。在-70 ℃下n=3.5时,产物为Nb2(AlH4)7;在-40 ℃下n=3.0时,产物为Nb2(AlH4)6;在20 ℃下n=2.5时,产物为Nb2(AlH4)5。
生成另一种氢化物LimNb(AlH4)n+m的反应如下:
NbCl5+(5+m)LiAlH4→
在-70 ℃下,产物为LiNb2(AlH4)7;在25 ℃下,产物为LiNb2(AlH4)5和LiNb(AlH4)3。
作者研究组曾尝试采用高压球磨法,在10 MPa的氢压下制备Nb2(AlH4)5(5.9% H2)。如图2所示,球磨产物中除了副产物LiCl之外,还出现了NbAl3和Al的衍射峰,说明Nb2(AlH4)5非常不稳定,在球磨过程中已经发生分解。到目前为止,Nb2(AlH4)5的分解放氢路径还未知,需要进一步研究。

图2 LiAlH4、NbCl5以及球磨NbCl5-5LiAlH4的XRD图谱Fig.2 XRD patterns of LiAlH4, NbCl5 and as-milled NbCl5-5LiAlH4samples
3.11 AgAlH4
AgAlH4的理论储氢量为2.9%。1952年,Wiberg等[26]通过AgClO4和LiAlH4在-80 ℃下乙醚溶液中反应首次制备出AgAlH4。当温度超过-50 ℃时,AgAlH4分解成Ag和Al,并释放出氢气,反应如下:
到目前为止,AgAlH4的结构特征尚未知,需要进一步研究。
3.12 TaH2(AlH4)2
TaH2(AlH4)2的理论储氢量为4.1%。1978年,Kost等[27]通过LiH、Al与TaCl5在乙醚溶液中反应生成Ta(AlH4)n。Ta(AlH4)n非常不稳定,会进一步分解形成TaH2(AlH4)2和AlH3。TaH2(AlH4)2是一种红色粉末,在温度超过130 ℃时发生分解。到目前为止,未见关于TaH2(AlH4)2形成过程以及分解放氢特性的进一步报道。
3.13 RE(AlH4)3(RE=La,Ce,Pr,Nd)
RE(AlH4)3(RE=La,Ce,Pr,Nd)的理论储氢量超过了5.1%。2009年,Weidenthaler等[43]尝试通过RECl3和NaAlH4采用机械球磨法制备RE(AlH4)3。在合成过程中,球磨罐内的压力升高(图3a),表明目标产物RE(AlH4)3已经发生分解。XRD结果表明,RE(AlH4)3在球磨过程中进一步分解为REAlxHy(图3b),合成及分解反应如下:
REAlxHy的成分与REAlH6非常接近,其晶体结构由孤立的[AlH6]3-八面体构成。以NdAlH6为例,REAlH6晶体结构沿不同晶面的投影如图3c和3d所示。Nd原子的氢原子配位数为12,其中6个氢原子在ab面,6个氢原子沿着c轴方向。REAlH6在加热过程中会进一步分解放氢。
除了LaAlH4,REAlH6(RE=Ce,Pr,Nd)的分解放氢过程均分2步进行(图3e)。对于Nd,Pr和Ce而言,100 ℃开始放氢,第一步放氢的峰值温度在170~180 ℃,第二步放氢过程则发生在180~270 ℃之间。LaAlH6的放氢过程发生在130~230 ℃之间,分解温度更高,放氢速度更快。DSC结果表明(图3f),Ce,Pr和Nd样品的第一步放氢过程是吸热反应,第二步放氢过程则是放热反应。放氢过程的原位XRD结果如图4所示[43],LaAlH6直接分解放氢形成LaAl4和Al,而Nd,Pr和Ce的中间氢化物REAlH6在分解过程会先形成REH3和Al,这2种产物在进一步加热过程中会反应生成REAl4,具体如下:

图3 REAlH6放氢过程的物相分析、结构示意图及分解过程曲线[43]:(a) 球磨过程的压力变化,(b) 球磨产物的XRD图谱,(c, d) NdAlH6晶体结构沿不同晶面的投影:[001] (c) 和[010]晶面 (d),(e) 放氢TPD曲线,(f) DSC曲线Fig.3 The dehydrogenation phase analysis, structural diagram and decomposition curves of REAlH6[43]: (a) hydrogen evolution during ball milling, (b) XRD patterns of as-milled products, (c, d) crystal structure of NdAlH6 in a projection along [001] (c) and along [010] (d), (e) TPD curves, (f) DSC curves
3.14 Eu(AlH4)2
Eu(AlH4)2的理论储氢量为3.7%。2012年,Pommerin等[32]采用EuCl2或EuCl3和NaAlH4通过机械球磨法制备了Eu(AlH4)2。如图5a所示,采用EuCl3时,球磨过程中发生如下反应:
EuCl3+NaAlH4→[EuCl2(AlH4)]+NaCl
采用EuCl2时,球磨过程生成Eu(AlH4)2,反应如下:
EuCl2+2NaAlH4→Eu(AlH4)2+2NaCl
在加热过程中,Eu(AlH4)2的分解放氢分2步进行:第一步发生在100~125 ℃,形成中间氢化物EuAlH5,第二步发生在200~225 ℃,进一步放氢形成EuAl4,这2步总的放氢量约为1.8%(图5b和5d)。随后在5,20,30和100 MPa的氢压下进行加氢反应,无法实现产物的再吸氢。
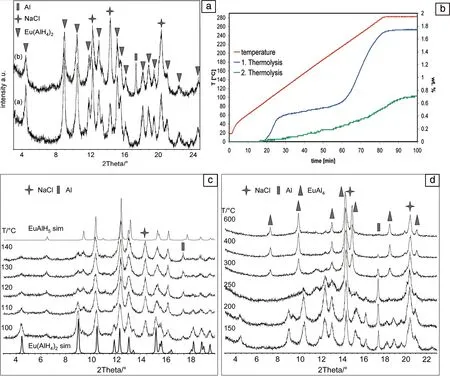
图5 Eu(AlH4)2球磨及放氢过程的物相分析及放氢曲线图[32]: (a) 球磨产物的XRD图谱;(b) 放氢过程的TPD曲线;(c,d) 不同温度下放氢产物的XRD图谱Fig.5 The dehydrogenation phase analysis and dehydrogenation curves of Eu(AlH4)2[32]: (a) XRD patterns obtained after ball-milling; (b) TPD curves of dehydrogenation process; (c,d) XRD patterns collected after dehydrogenation at different temperatures
3.15 Yb(AlH4)2和Yb(AlH4)3
Yb(AlH4)2和Yb(AlH4)3的理论储氢量分别为3.4%和4.5%。1978年,Kost等[25]通过Yb的卤化物和LiAlH4在-110 ℃下乙醚溶液中反应首次合成出Yb(AlH4)2。研究表明,Yb(AlH4)2为非晶态,起始放氢温度为70 ℃。结合氢化物在-110~25 ℃之间的放氢量,他们提出Yb(AlH4)2的放氢路径如下:
最近,作者研究组首次采用高压球磨法在10 MPa氢压下成功制备出Yb(AlH4)3,并系统研究了其放氢反应机制、放氢性能和可逆性[28]。研究表明,产物中Yb(AlH4)3呈现非晶态(图6a)。TGA-DSC-MS联合表征的结果显示,Yb(AlH4)3的放氢过程分4步进行,起始放氢温度为100 ℃,在100~400 ℃之间的放氢量为4.44%(图6b)。不同温度下放氢产物的XRD分析结果表明,Yb(AlH4)3的放氢过程反应路径如下(图6c):
Yb(AlH4)3→YbAlH6+2Al+3H2
第一步放氢过程的等温放氢结果表明(图6d),Yb(AlH4)3在160 ℃下的放氢量为2.3%,约20 min即可完成90%放氢量,且第一步放氢反应的表观活化能为99.6 kJ/mol。在160 ℃、14 MPa的条件下进行再吸氢实验,产物为YbHCl,无法实现Yb(AlH4)3可逆吸氢。
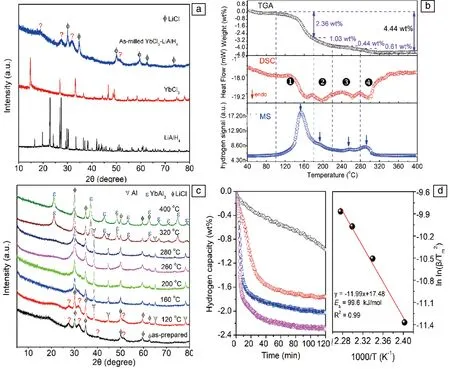
图6 Yb(AlH4)3-3LiCl放氢过程的物相分析及放氢曲线[28]: (a) 球磨产物的XRD图谱,(b) 放氢TGA-DSC-MS曲线,(c) 不同温度下放氢产物的XRD图谱,(d) 等温放氢及动力学拟合曲线Fig.6 The dehydrogenation phase analysis and dehydrogenation curves of Yb(AlH4)3-3LiCl[28]: (a) XRD patterns obtained after ball-milling, (b) TGA-DSC-MS curves, (c) XRD patterns of dehydrogenated samples at different temperatures, (d) isothermal dehydrogenation curves and Kissinger plot fitting curve
3.16 Th2AlH4
到目前为止,尚未发现金属Th与官能团[AlH4]-形成配位氢化物的报道,但Th2Al合金能够与氢反应能够生成金属间氢化物Th2AlH4[44]。Sørby等[45]将Th2Al合金在450 ℃真空下活化,然后在0 ℃下起始压力为0.015 MPa时进行吸氘,能够形成Th2AlDx(x=2.3,2.7,3.9)。
Vajeeston等[46]通过理论研究,发现Th2AlH4的晶体结构属I4/mcm空间群。如图7所示,Th2Al的晶体结构中包含4种不同的间隙位置,分别为16l、4b、32m和16k,均可以作为储氢位点。吸氘时,结构中的16l位点被完全占据,5个Th原子形成的2个四面体间隙中包含2个氢原子,且Th2AlD4的结构完全有序。到目前为止,未见关于Th2AlH4分解特性和储氢性能的进一步报道。

图7 Th2AlH4的晶体结构[46]Fig.7 The crystal structure of Th2AlH4[46]
4 结 语
过渡金属配位铝氢化物的研究起步很早,但目前多数此类氢化物的结构特征及储氢性能仍然未知。本文首先介绍了过渡金属配位铝氢化物的2种制备方法。采用低温液相法制备过程中,有机溶剂会和氢化物形成较为稳定的络合物,结构极其复杂,难以解析,同时氢化物在加热放氢过程中混有大量的有机物。高压球磨法能够制备一些室温下相对稳定的氢化物,但产物中混有盐类副产物,降低了体系的有效储氢量。其次,简要介绍了过渡金属配位铝氢化物的制备条件及形成机理,重点总结了在室温下相对稳定的氢化物TM(AlH4)n(TM=Y, Yb, Eu,RE)的结构、性能表征和储氢机理等,但这些氢化物普遍存在着吸放氢速率缓慢、可逆程度低、可逆条件苛刻等问题。多数氢化物的热力学稳定性非常低,目前的相关研究工作尚少,其储氢特性及机理研究仍处于初步探索阶段。鉴于上述问题,结合作者长期的研究体会,建议在以下3方面进行系统性的基础研究,为探索高容量储氢材料提供重要参考:
(1)通过理论研究[46],建立过渡金属阳离子与过渡金属配位铝氢化物热力学稳定性之间的关系,为判断氢化物的稳定性提供理论指导。
(2)开发新的合成和表征技术,在低温下实现氢化物合成和吸放氢表征,为表征氢化物和解决储氢问题提供新方法。
(3)开发新的热力学调控策略,提高过渡金属配位铝氢化物的热力学稳定性,开发室温下具备可逆储氢能力的氢化物,拓展过渡金属配位铝氢化物的应用。
